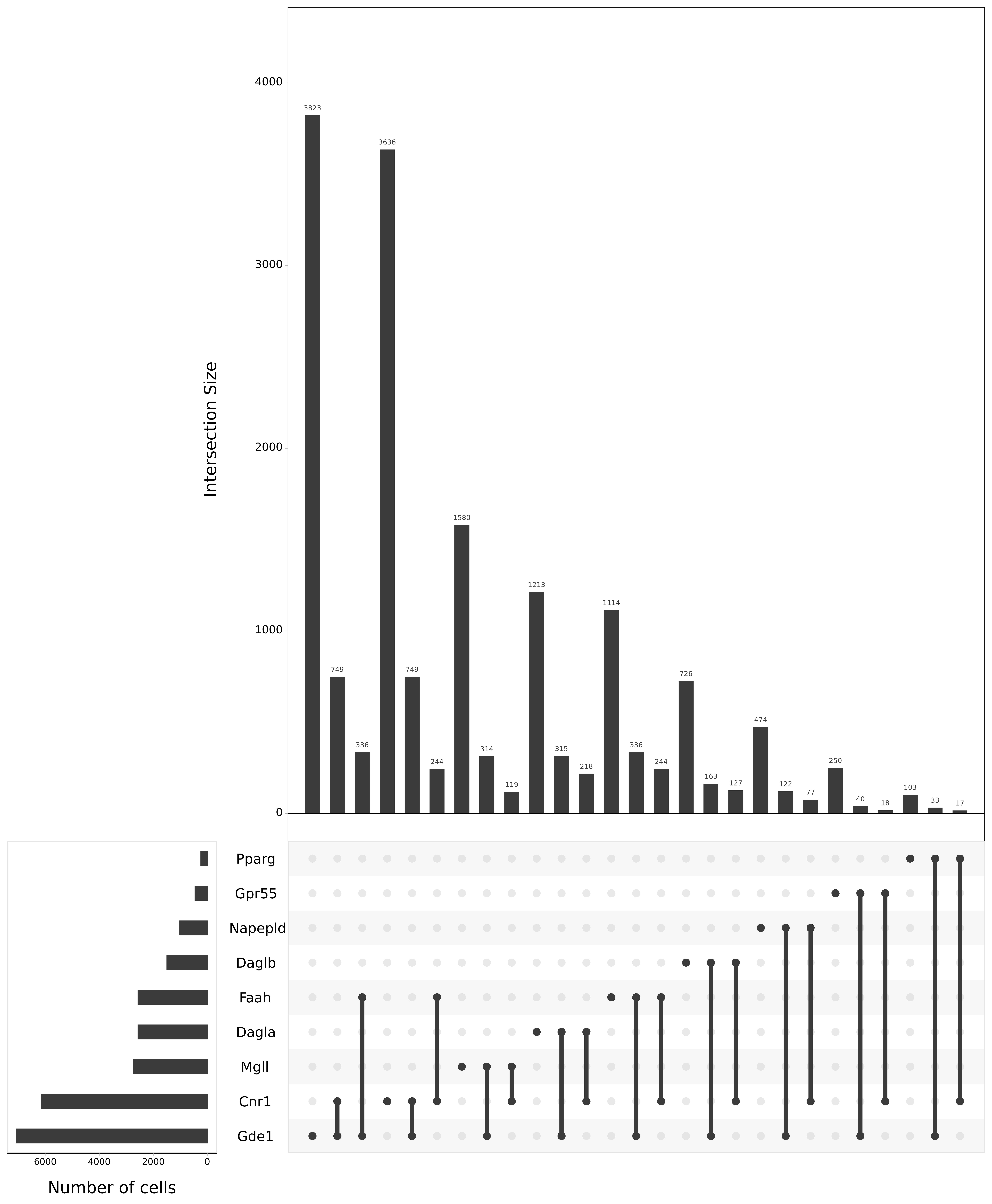
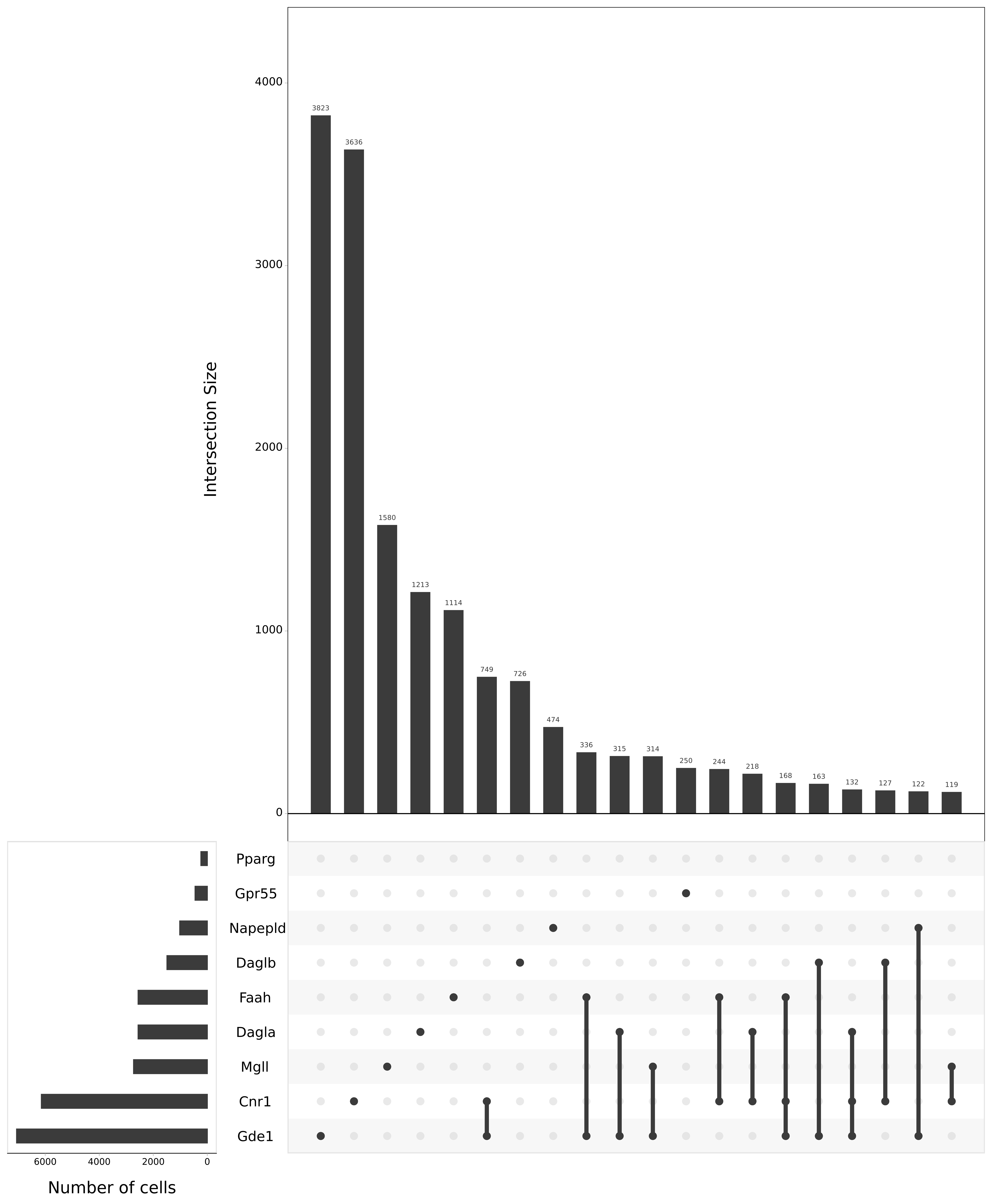
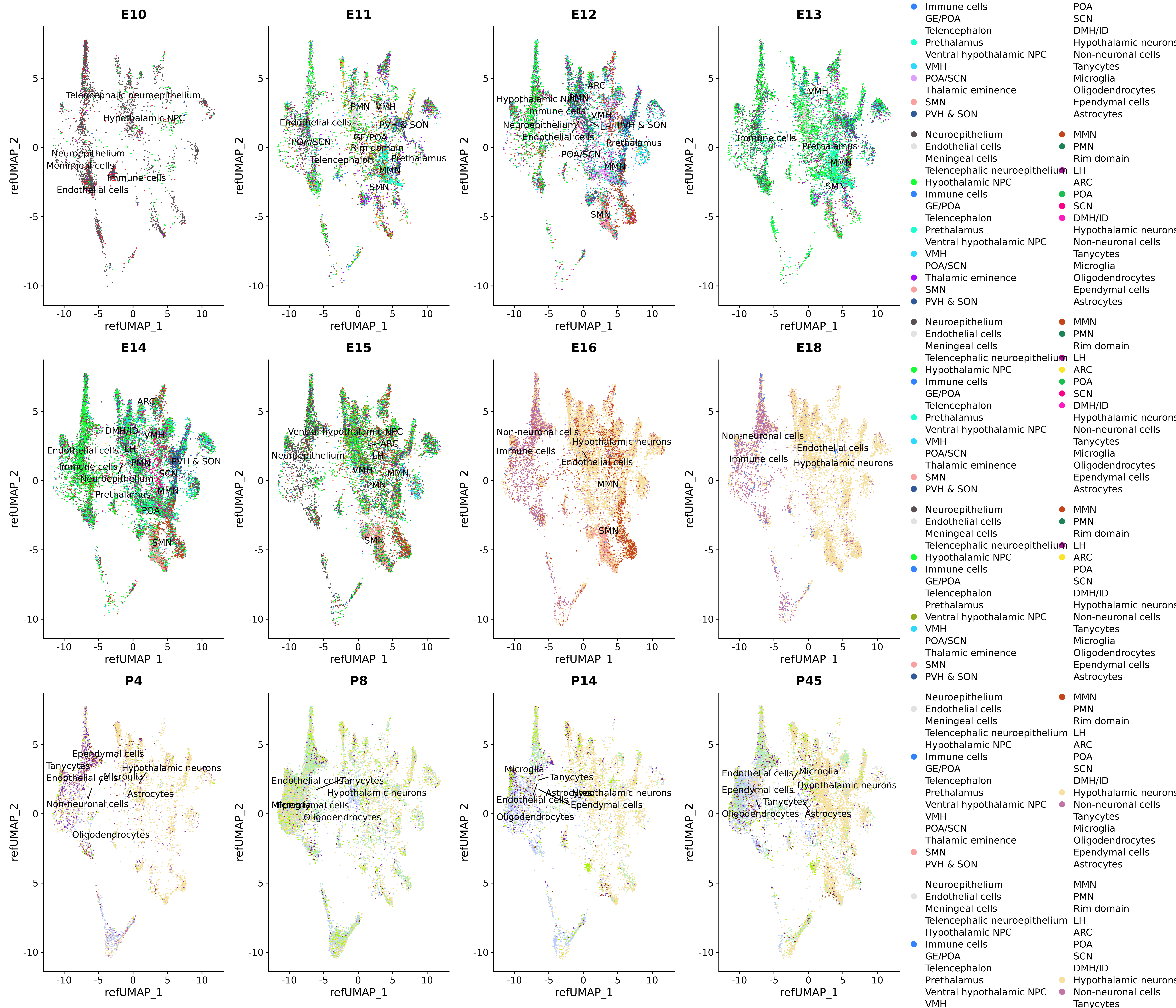
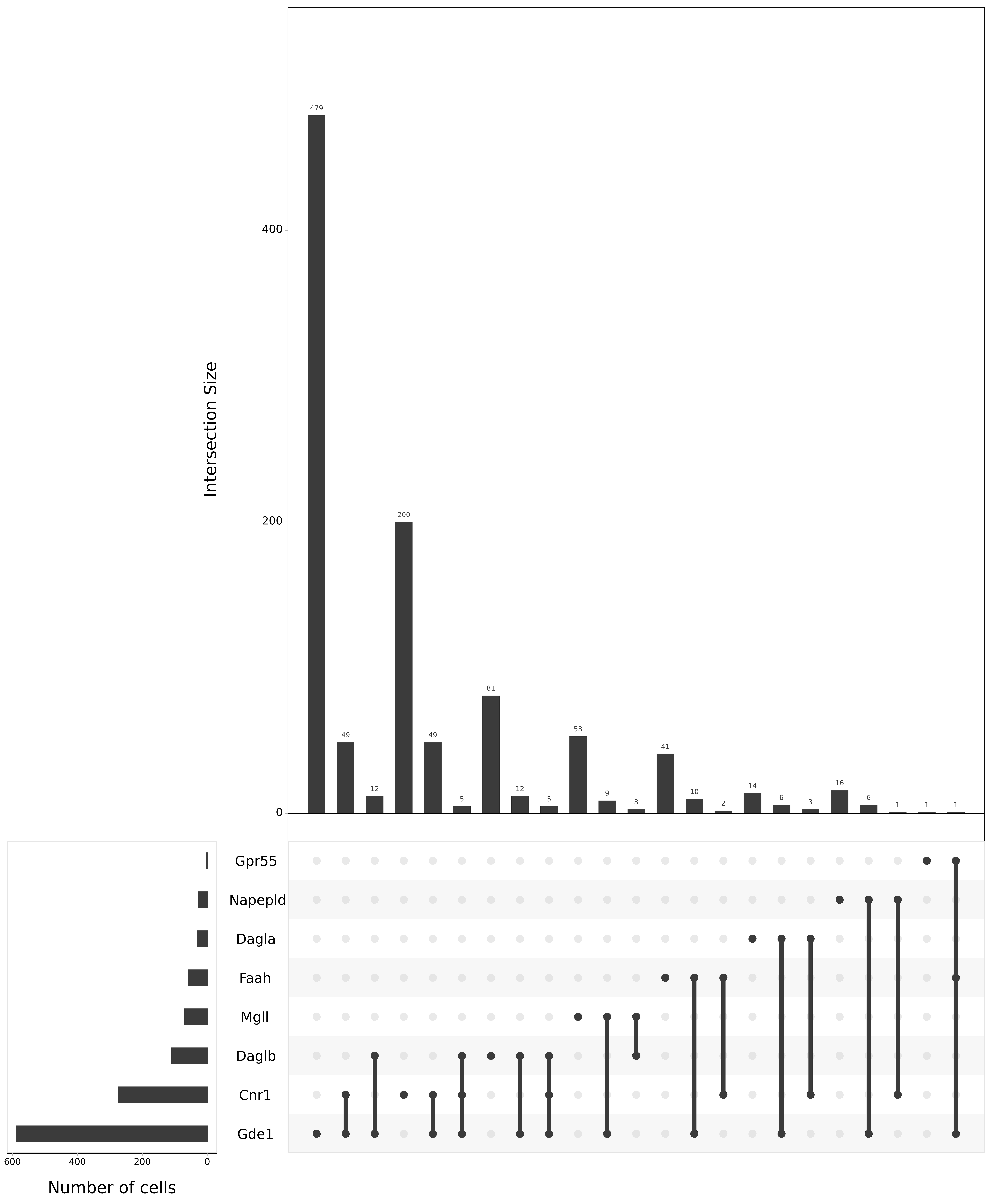
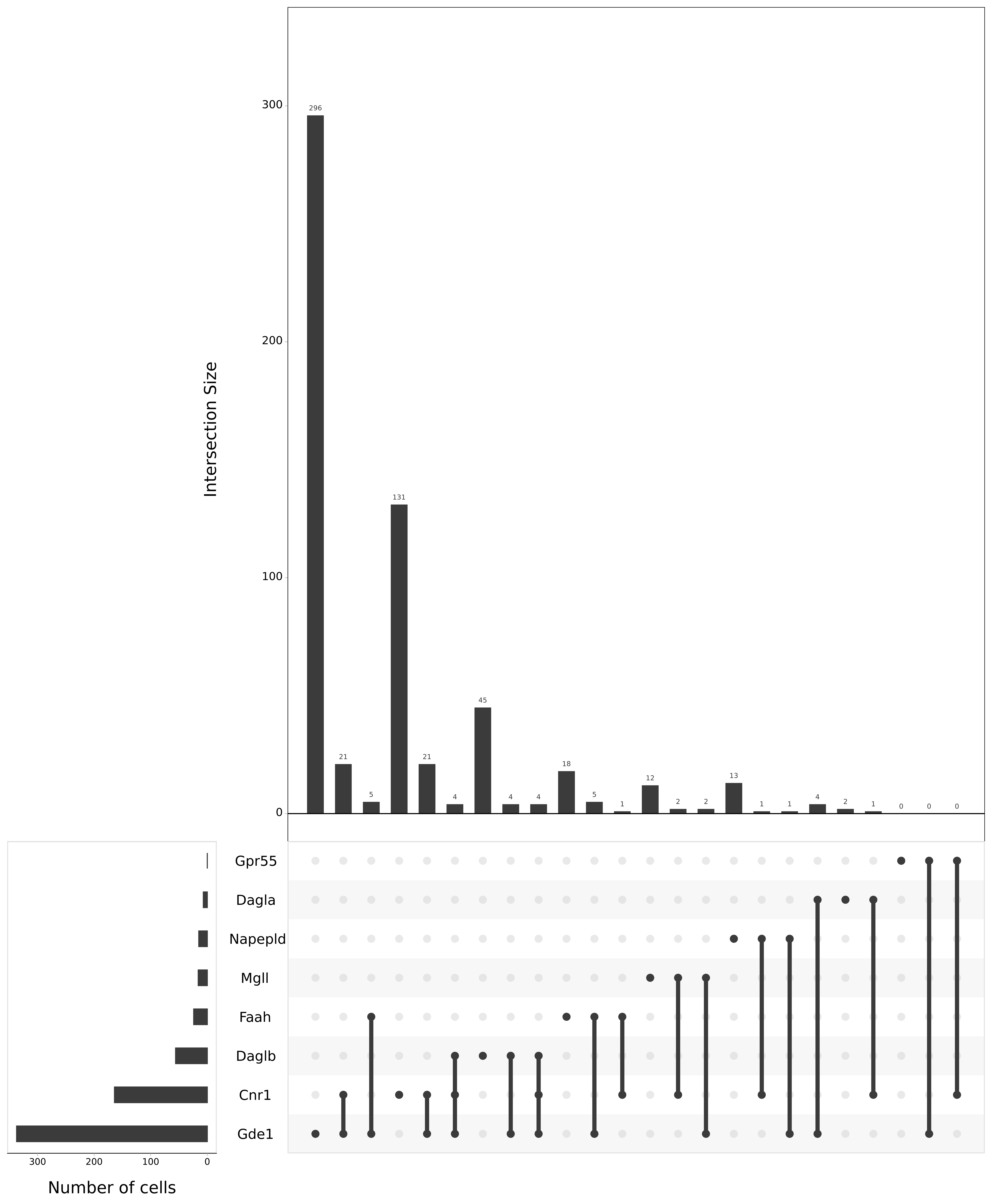
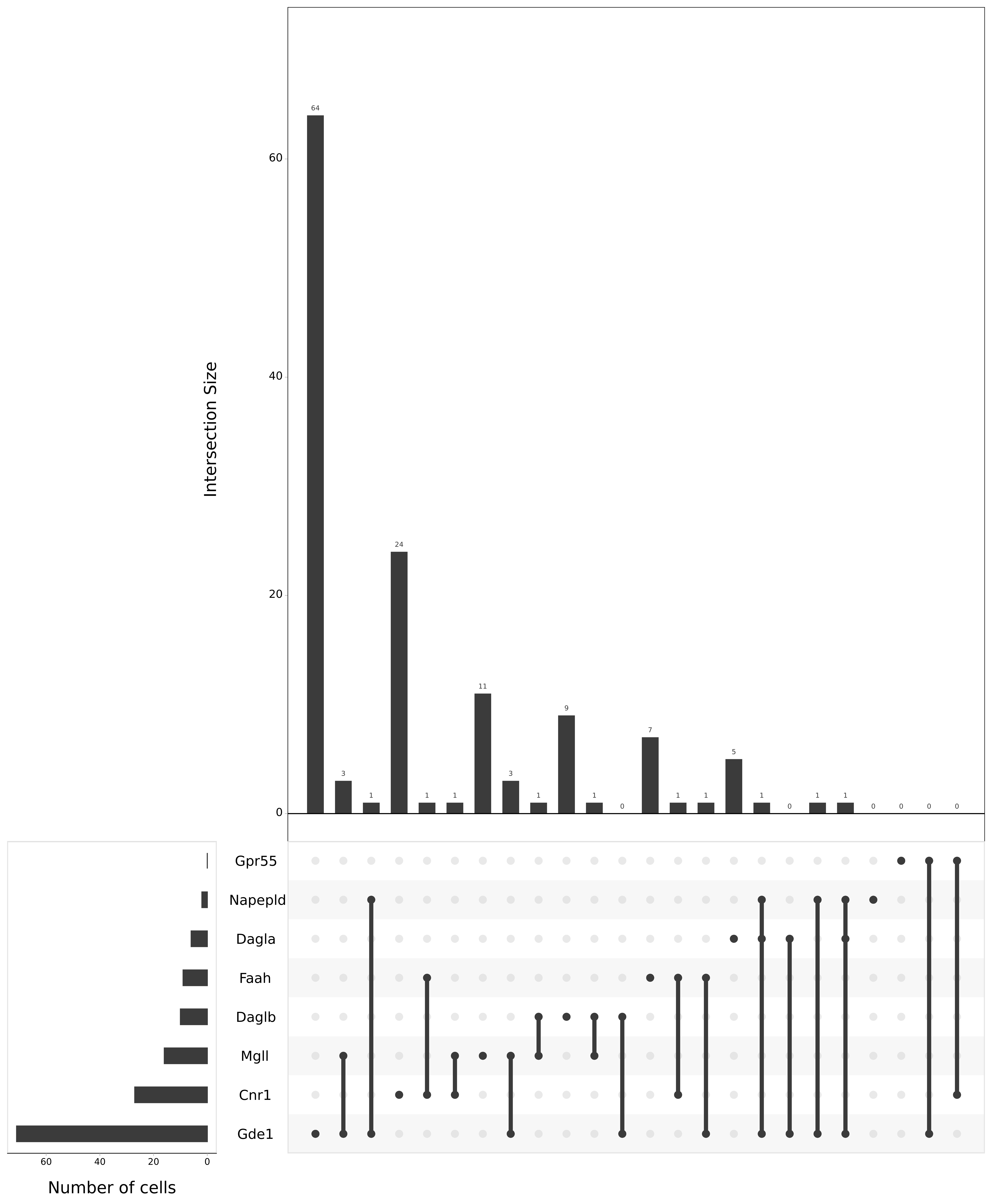
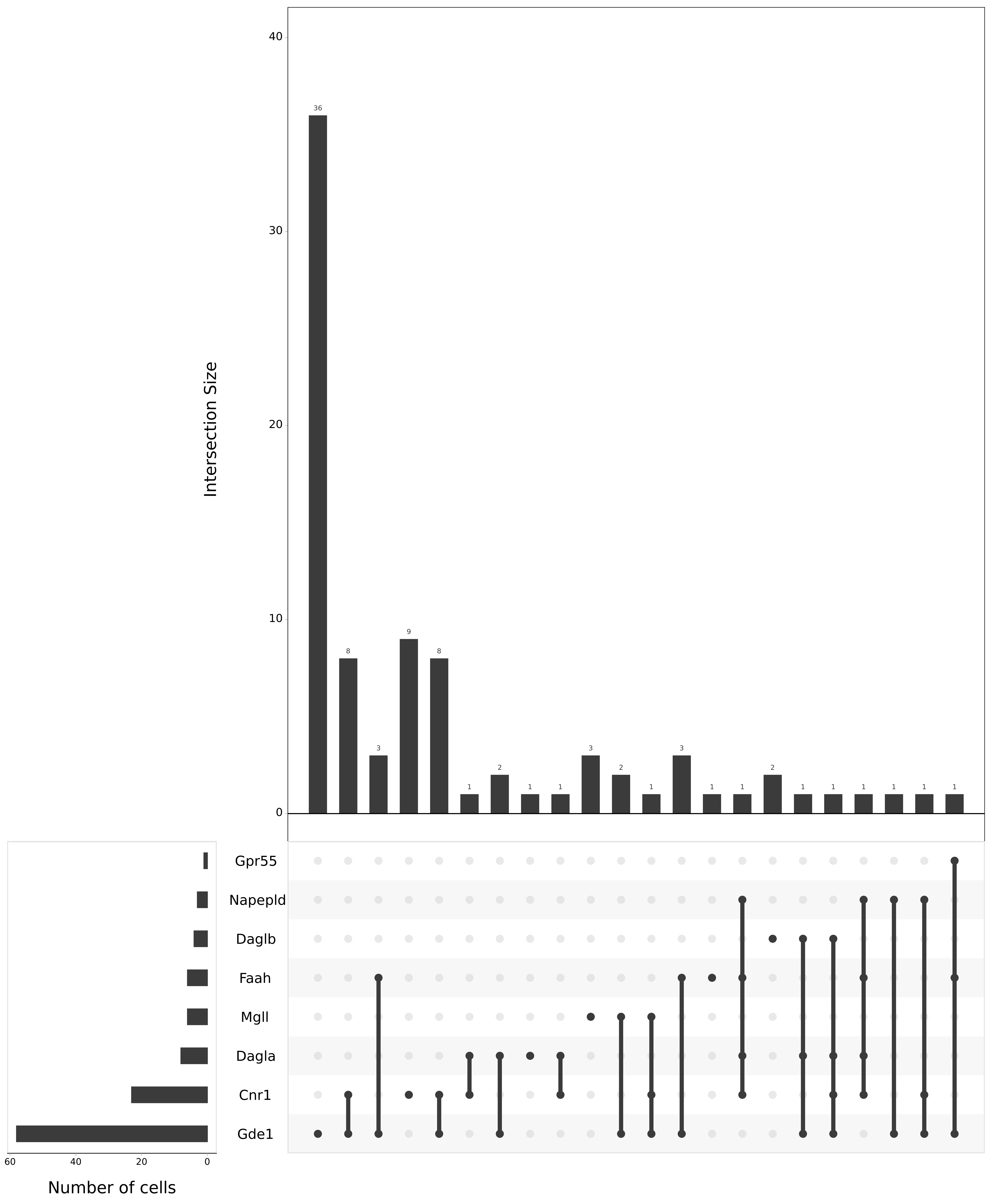
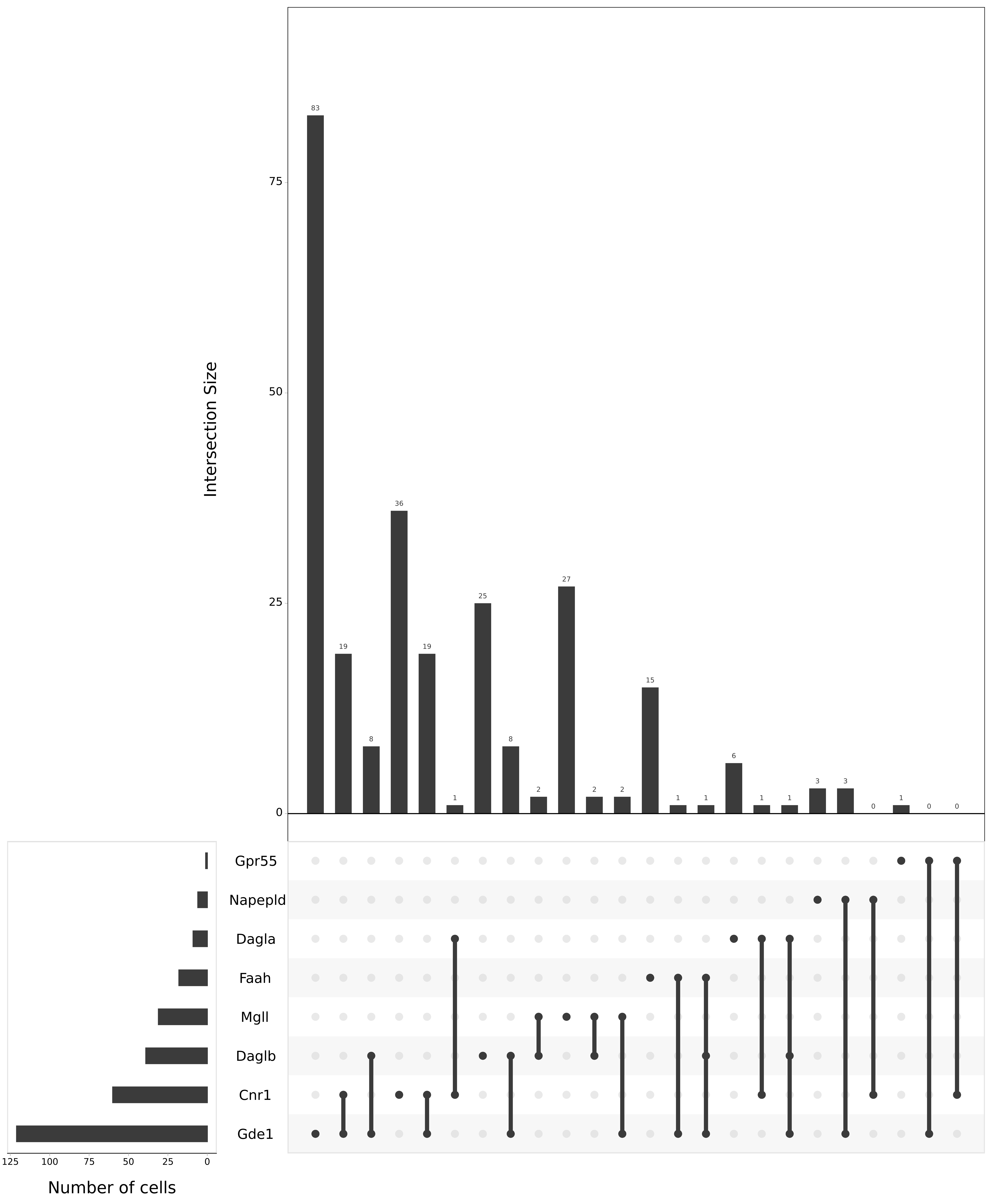
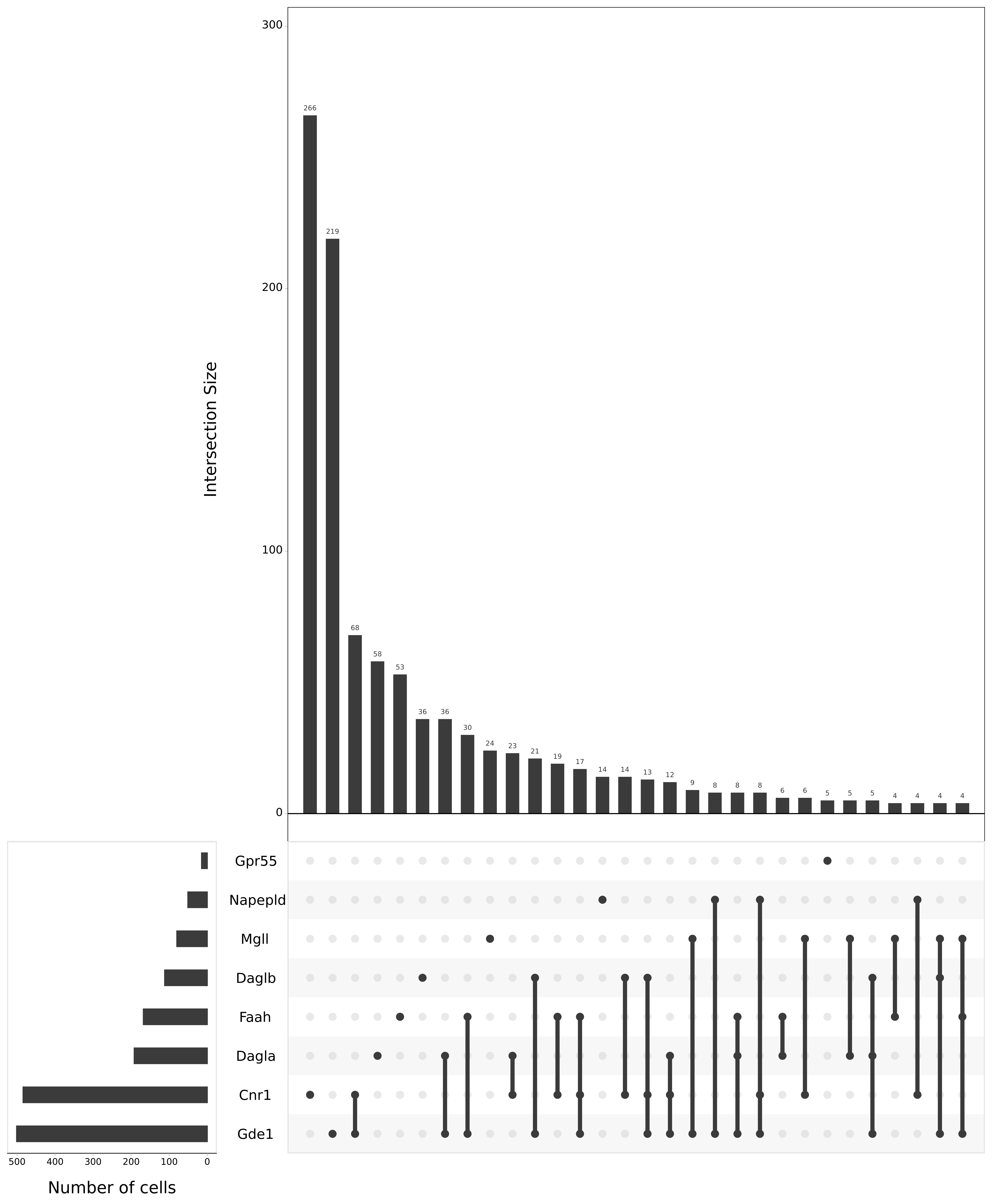
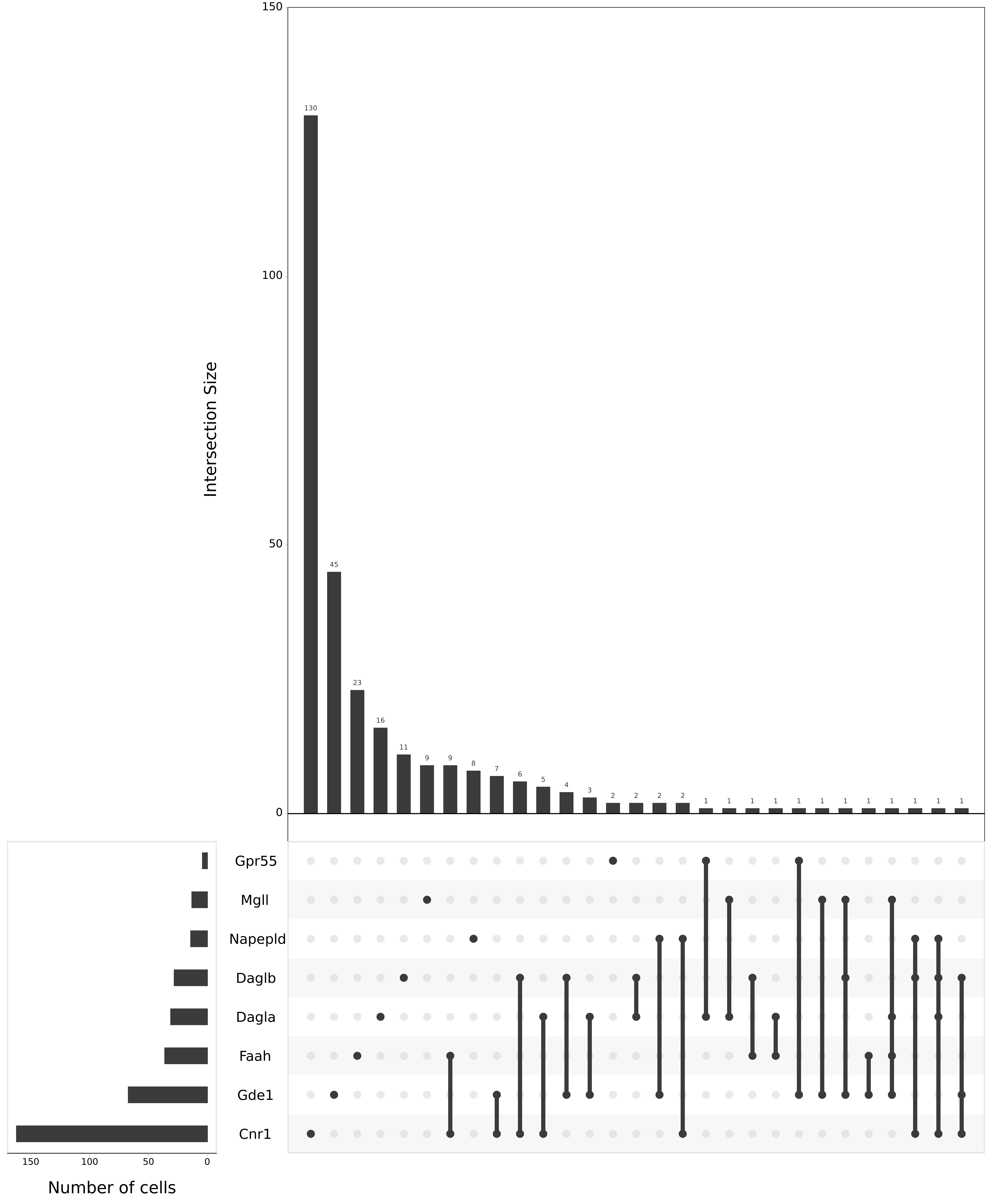
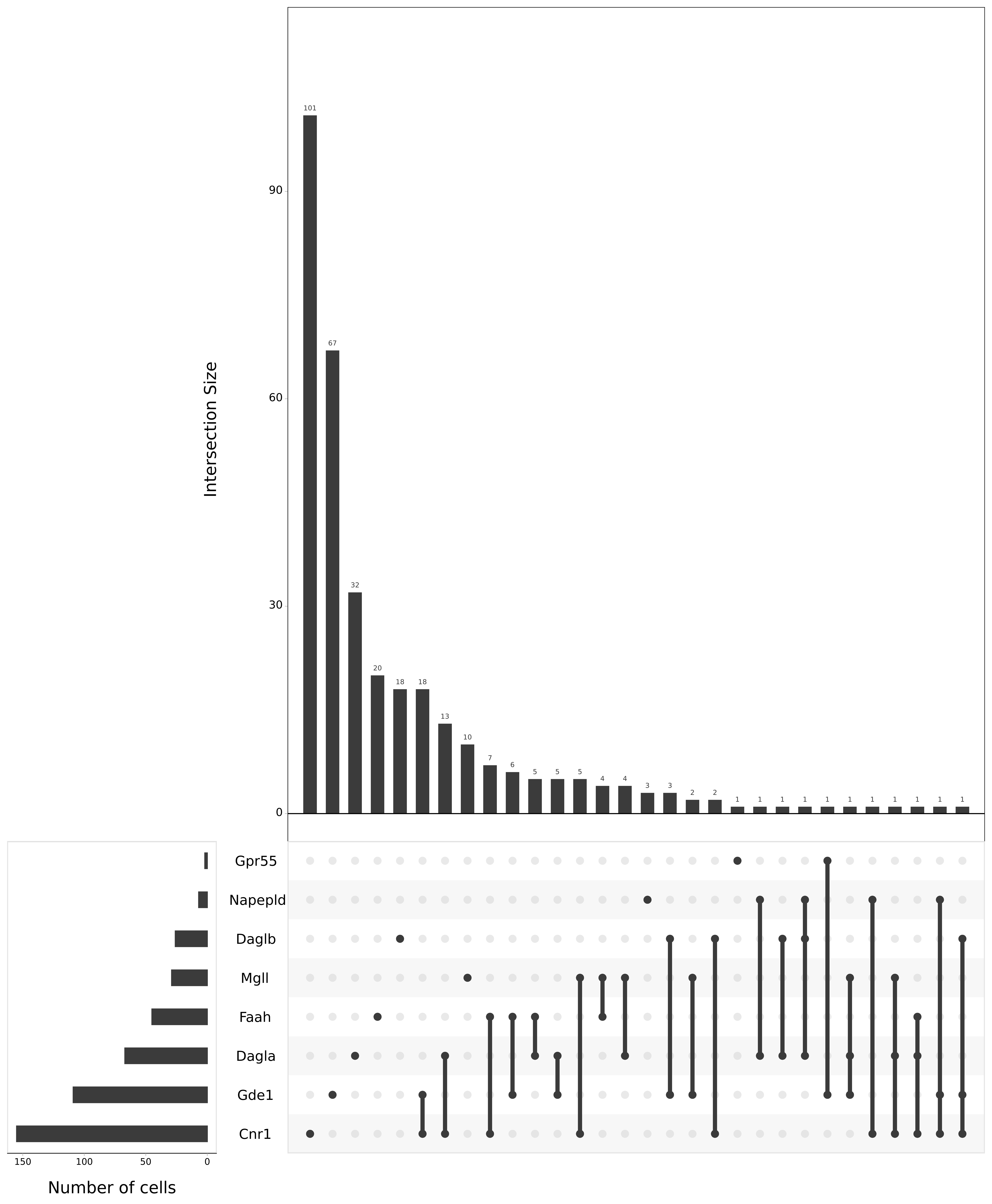
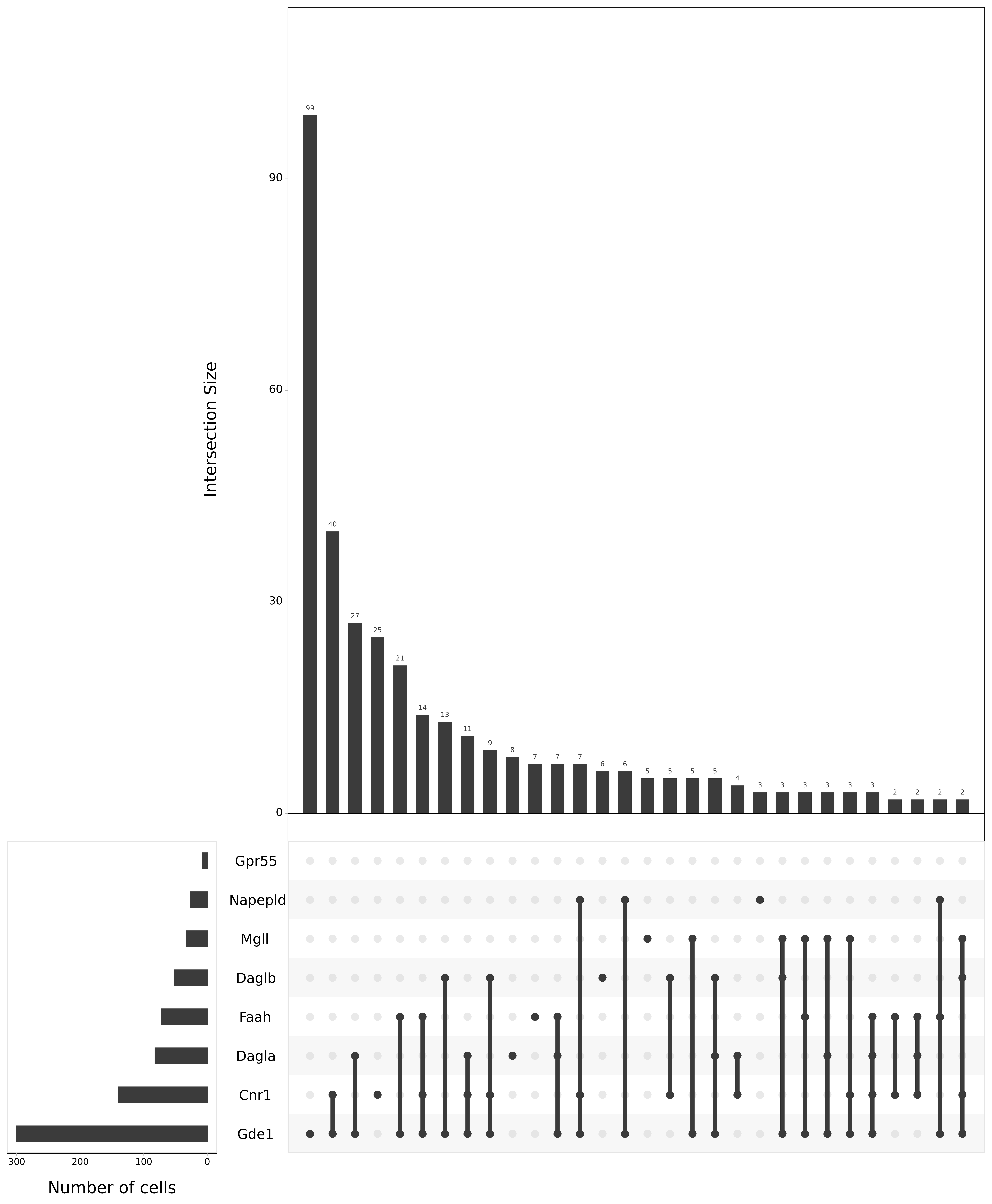
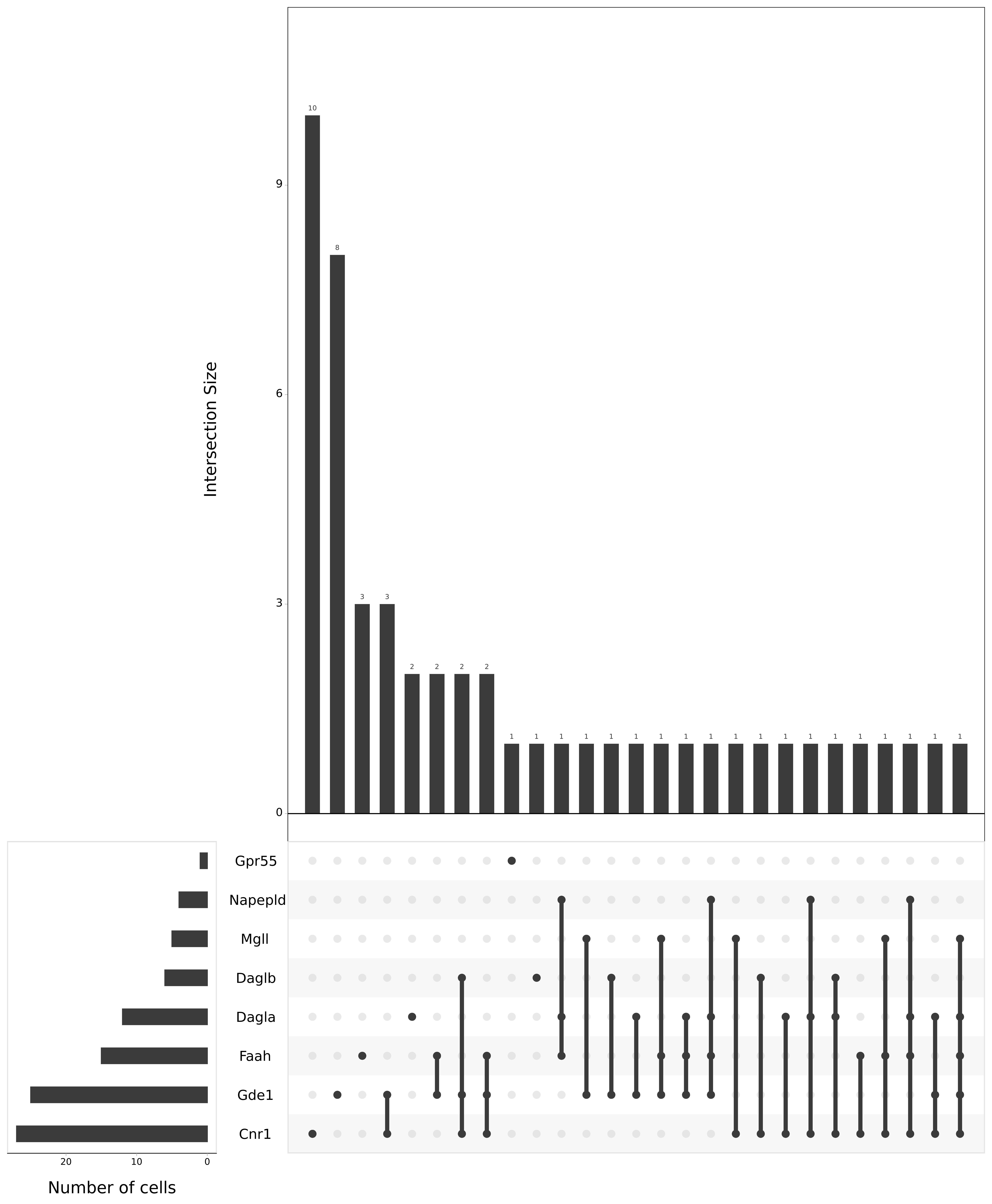
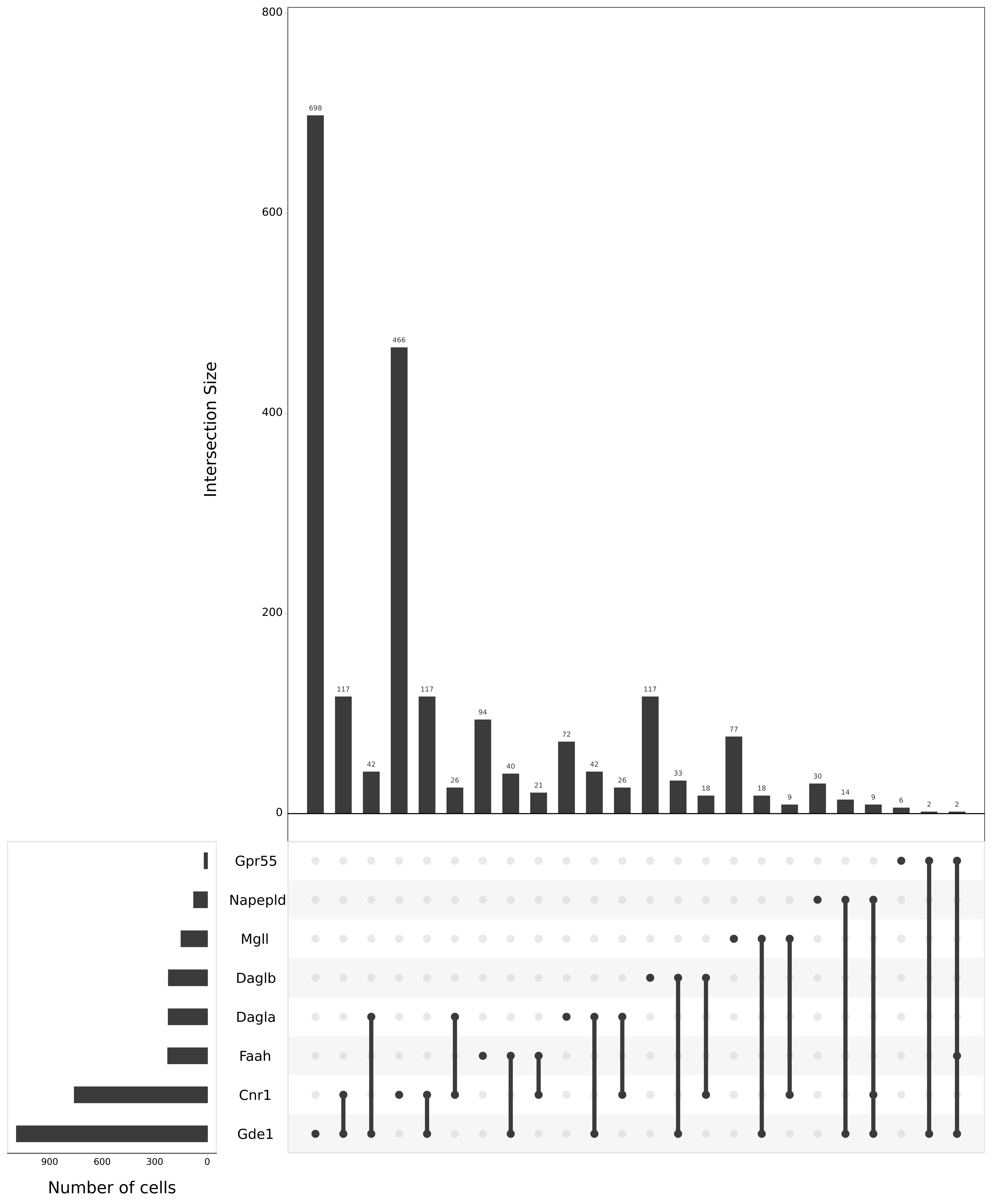
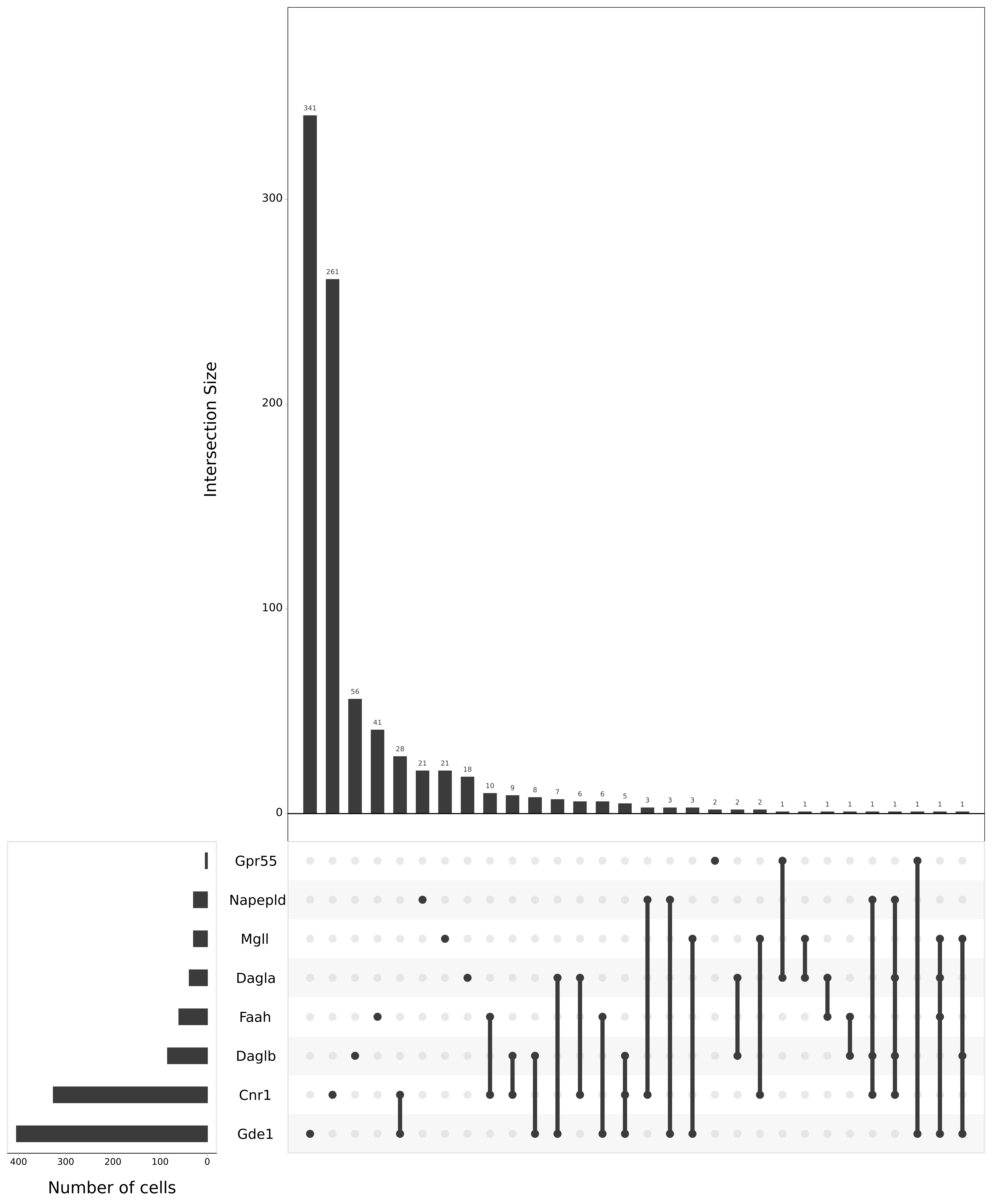
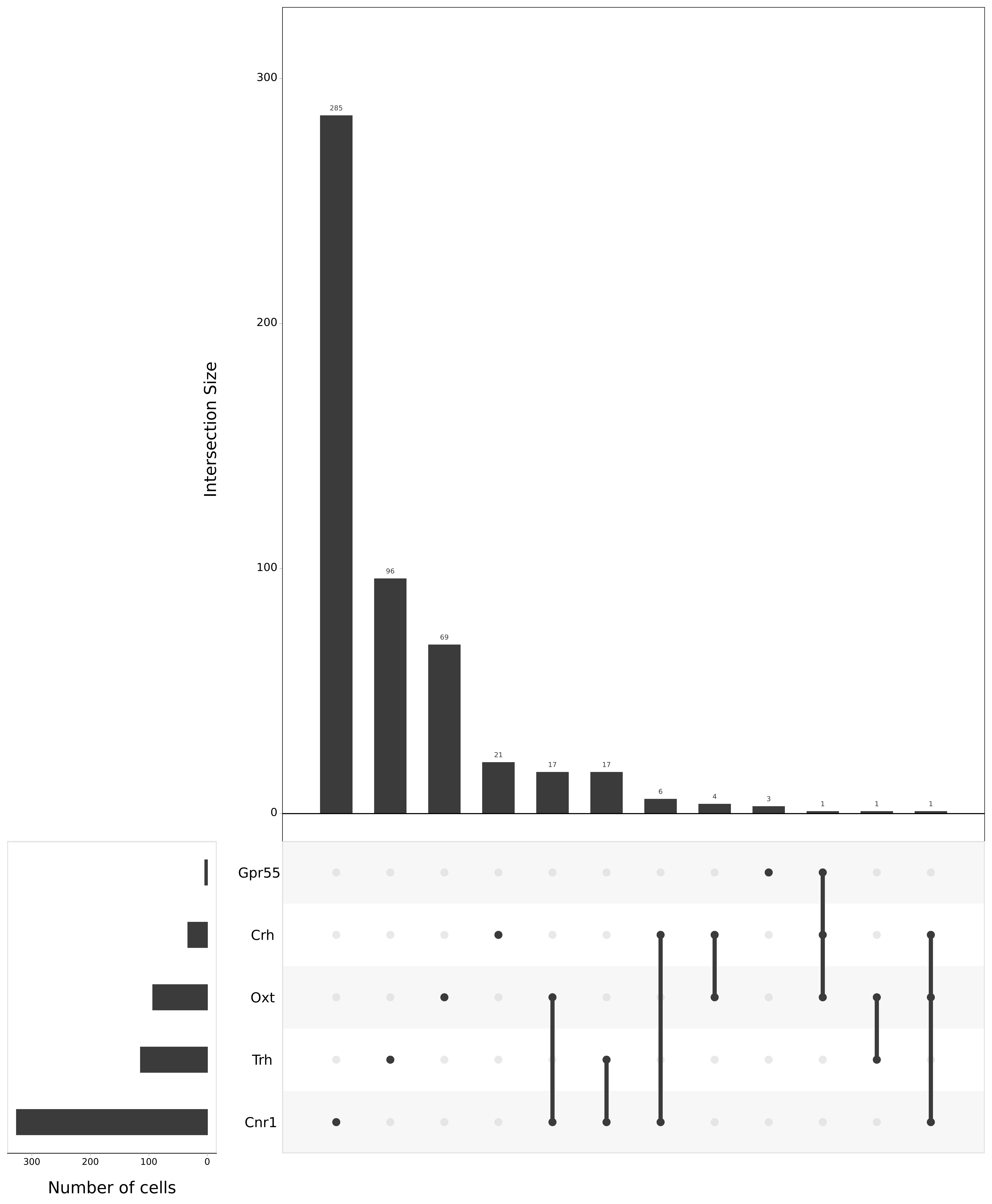
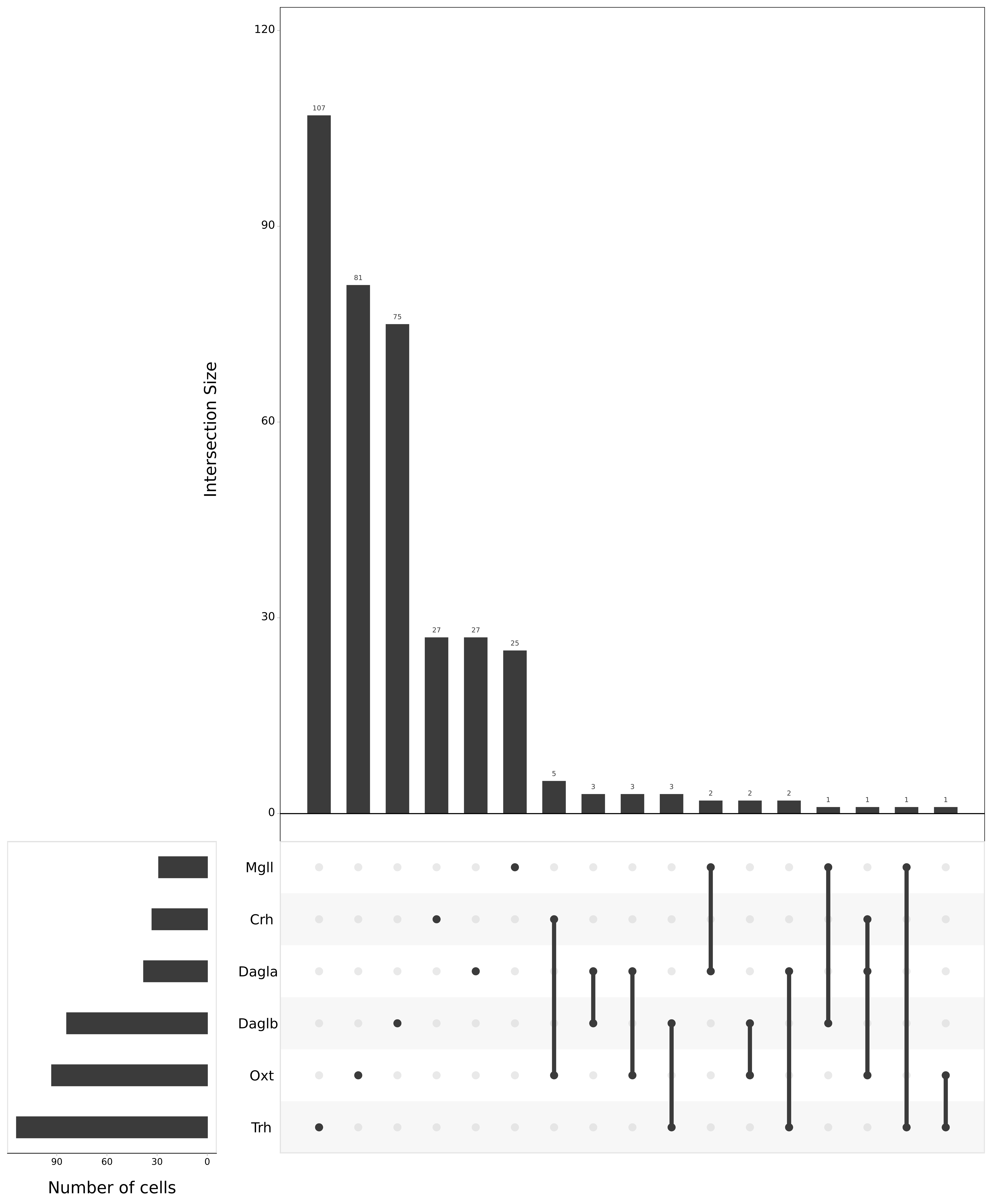
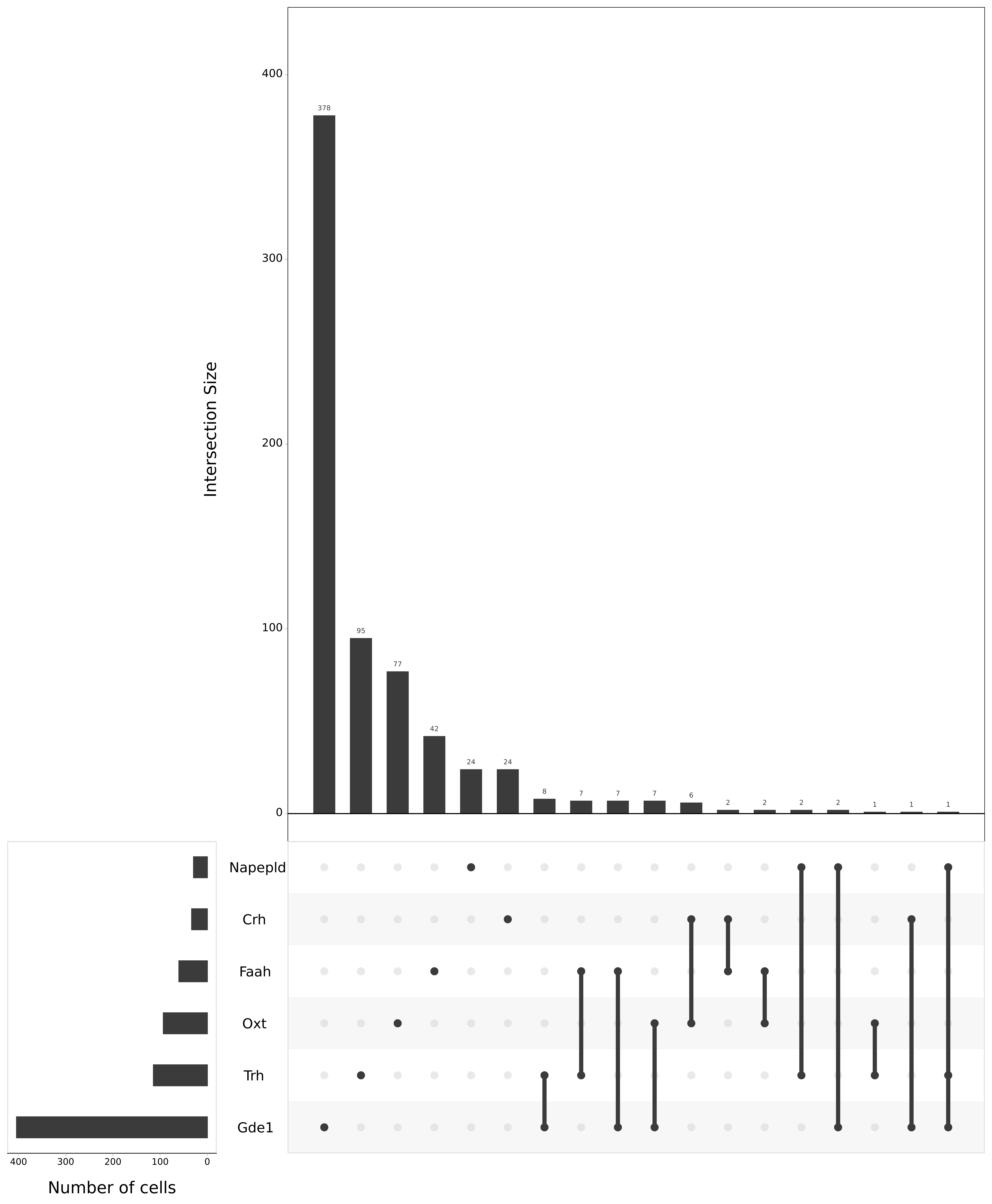
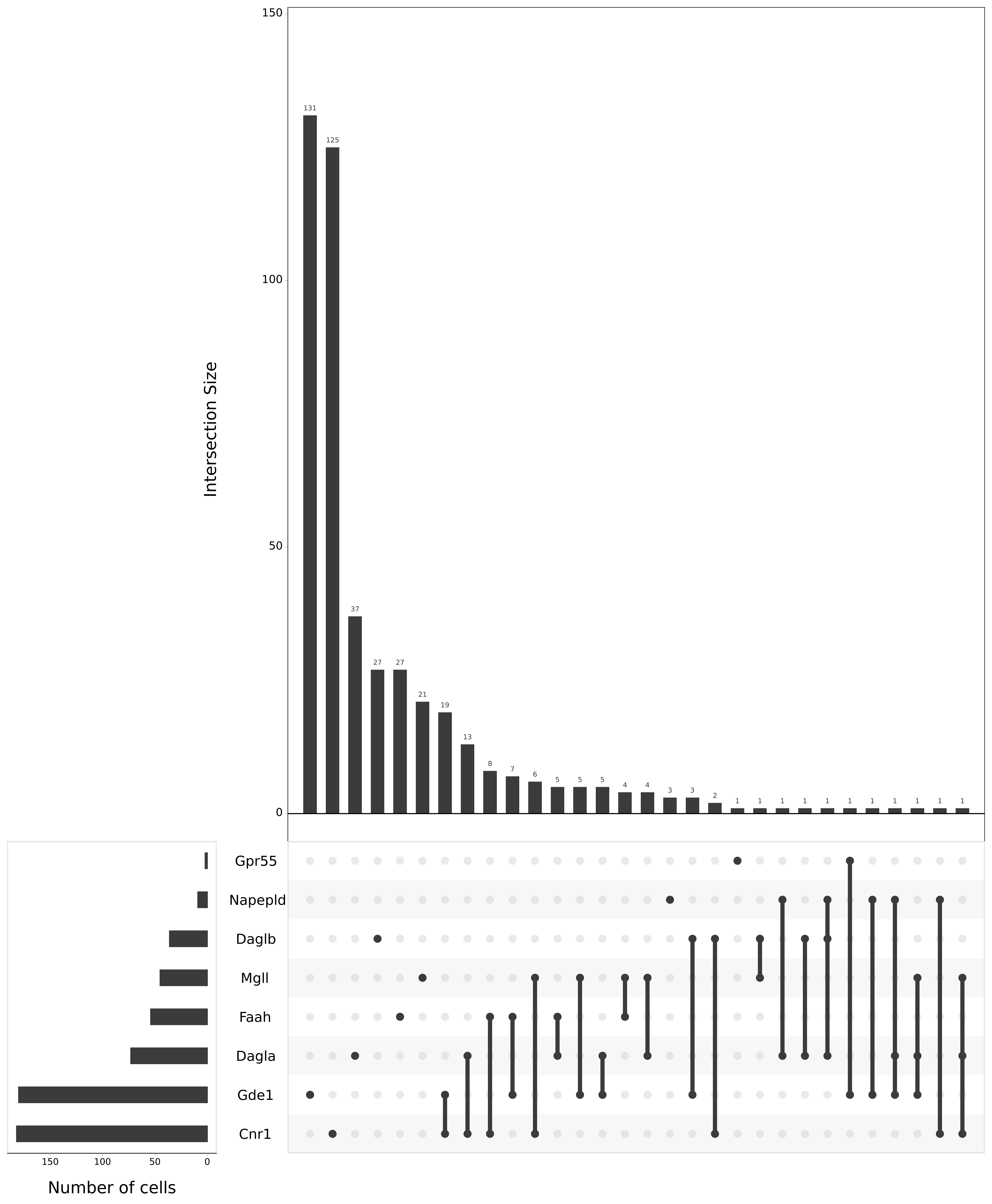
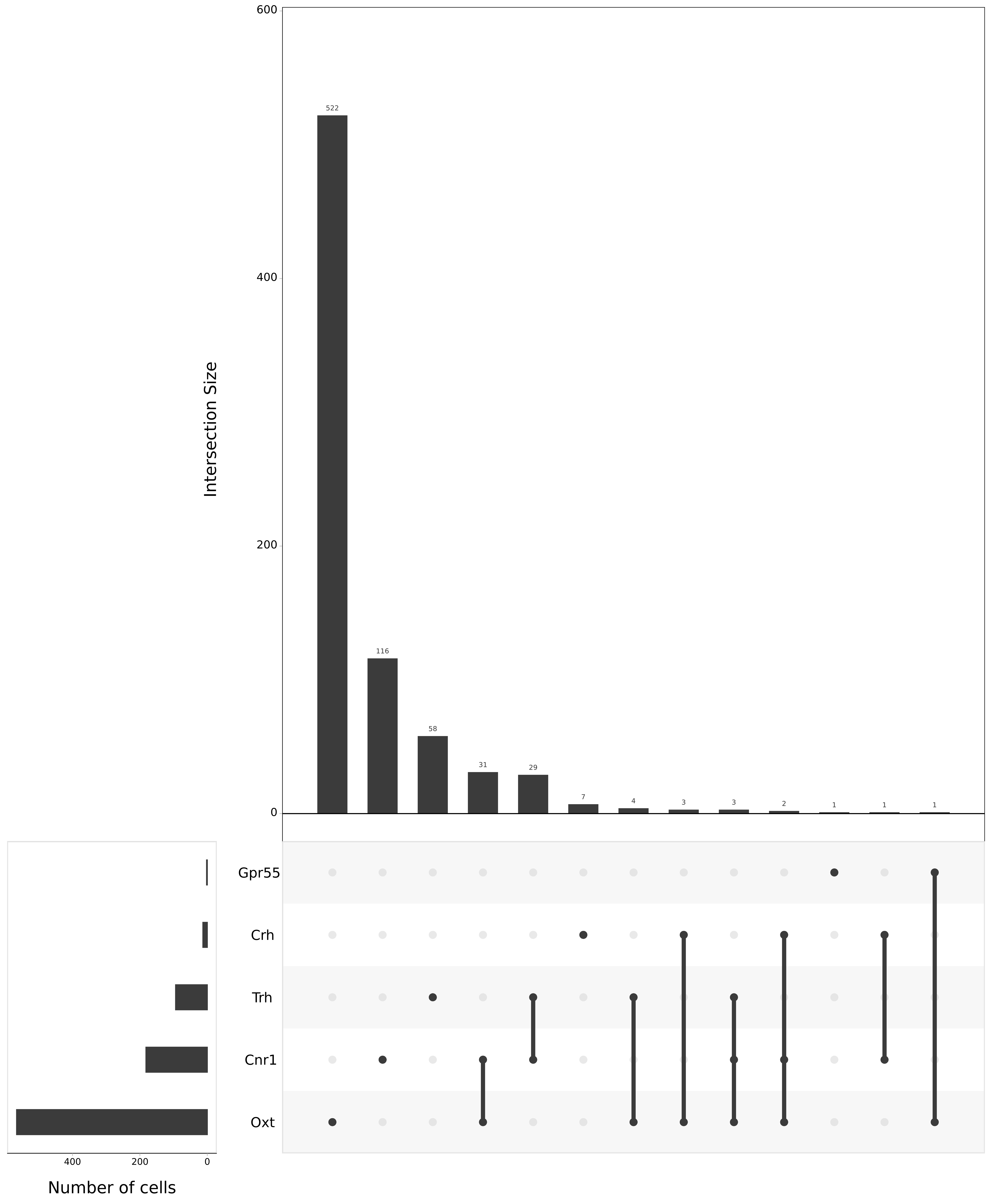
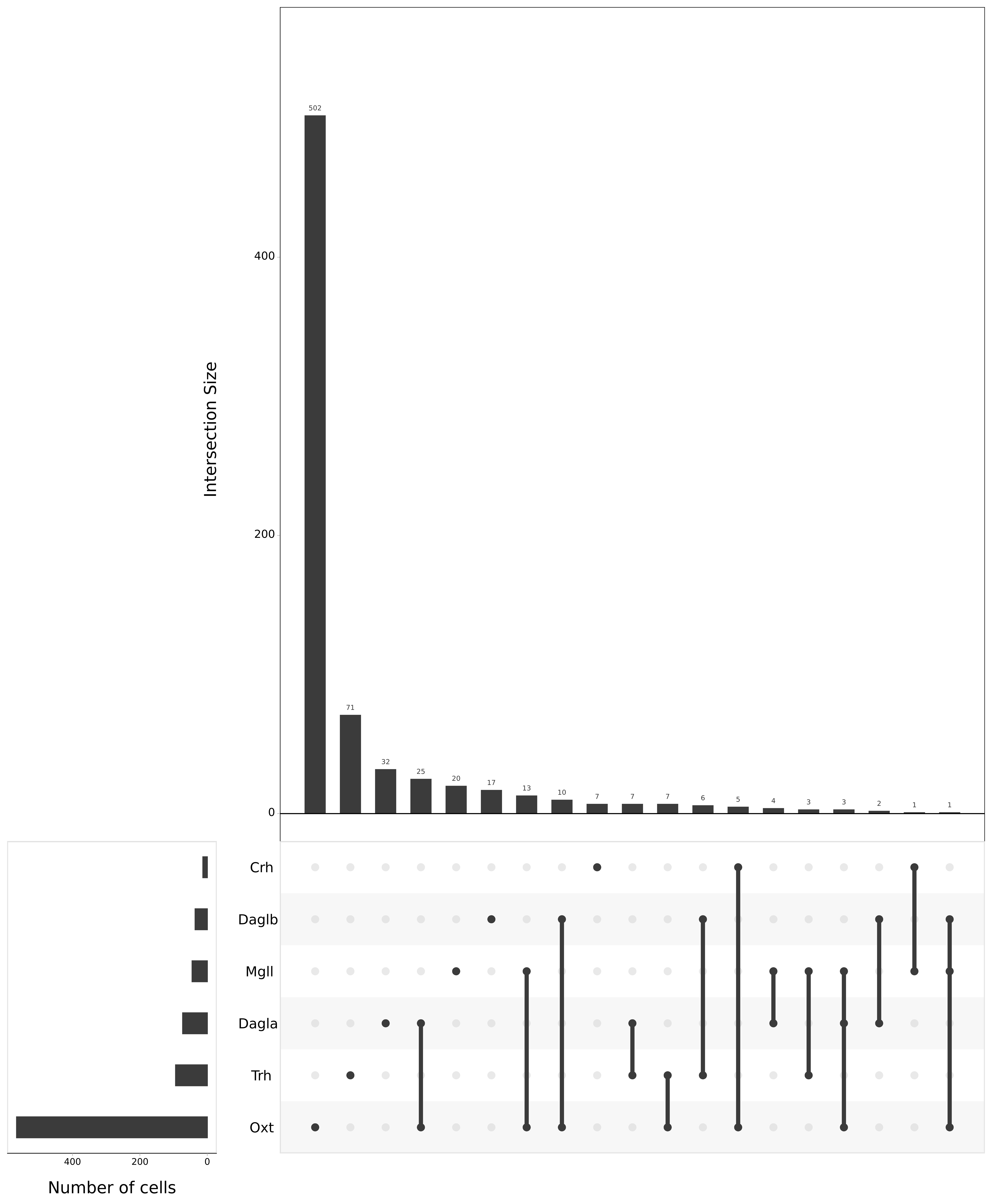
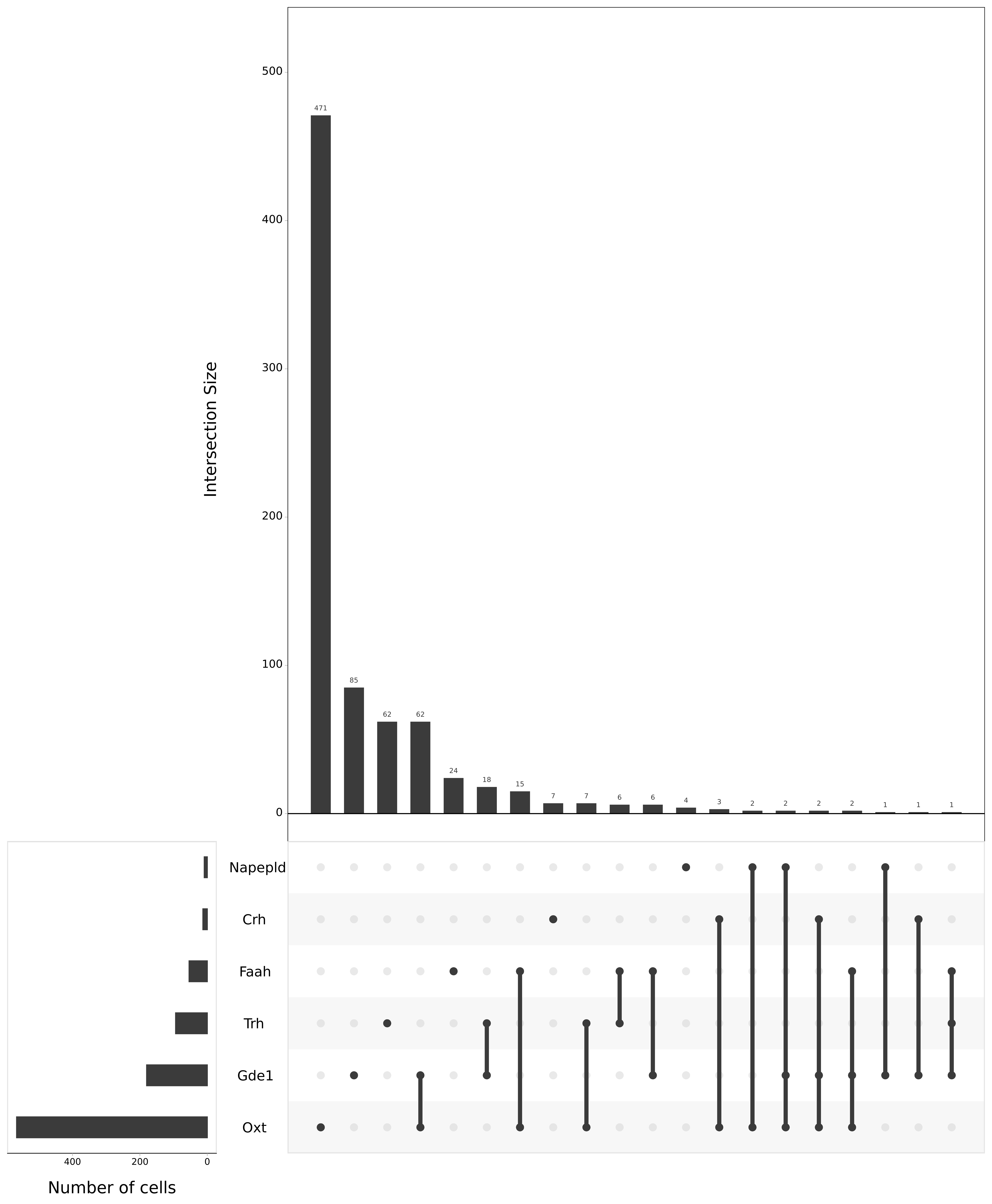
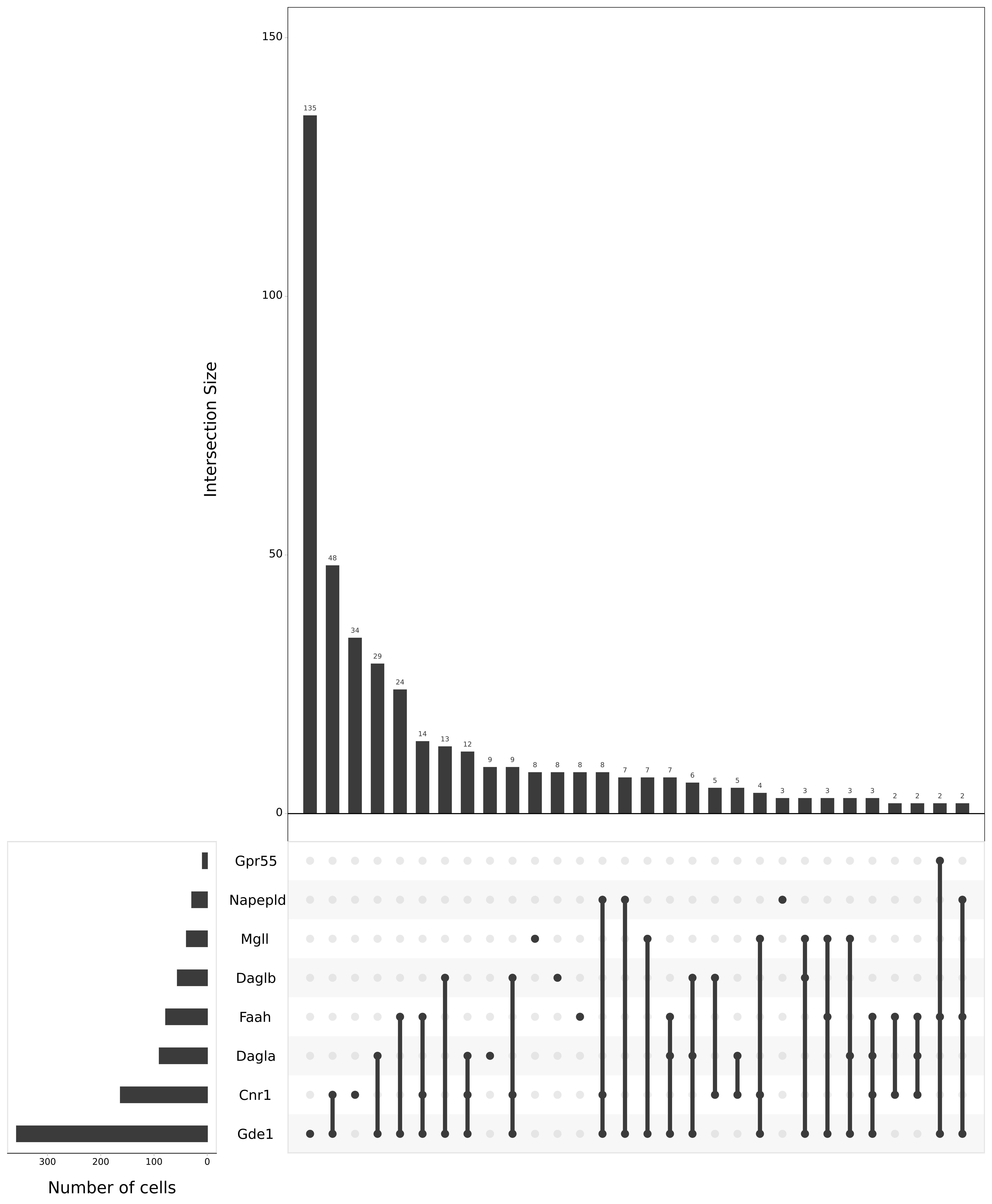
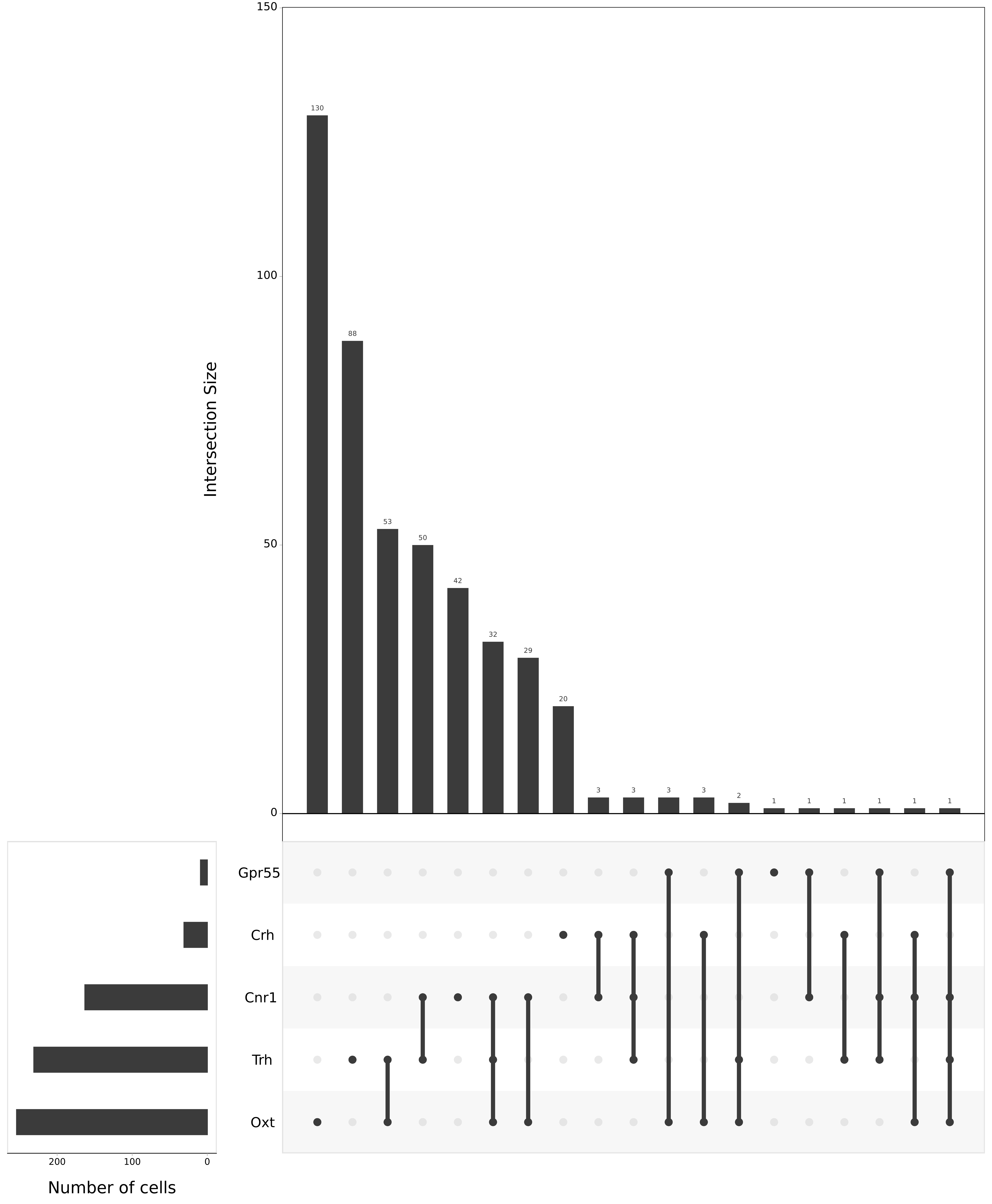
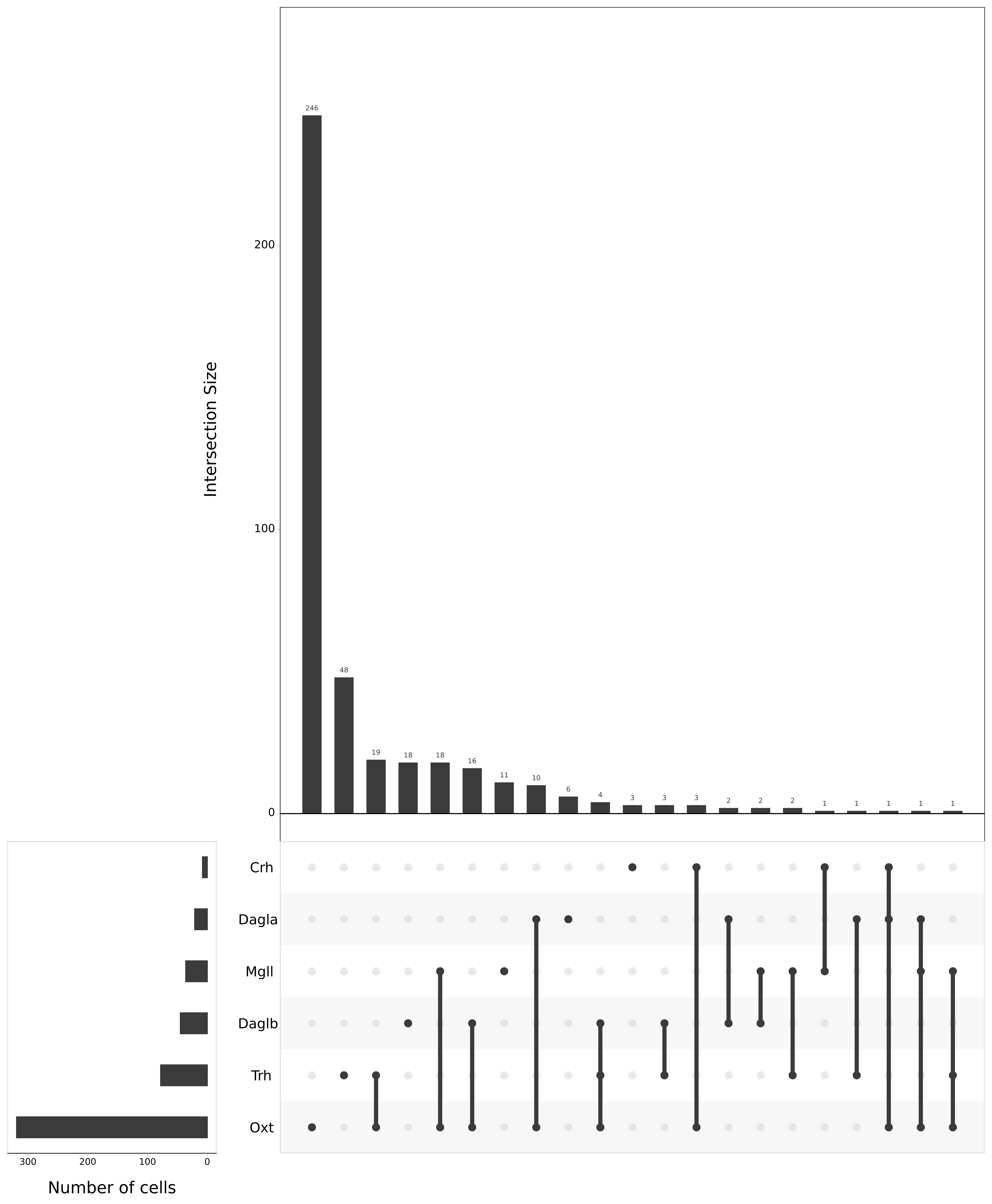
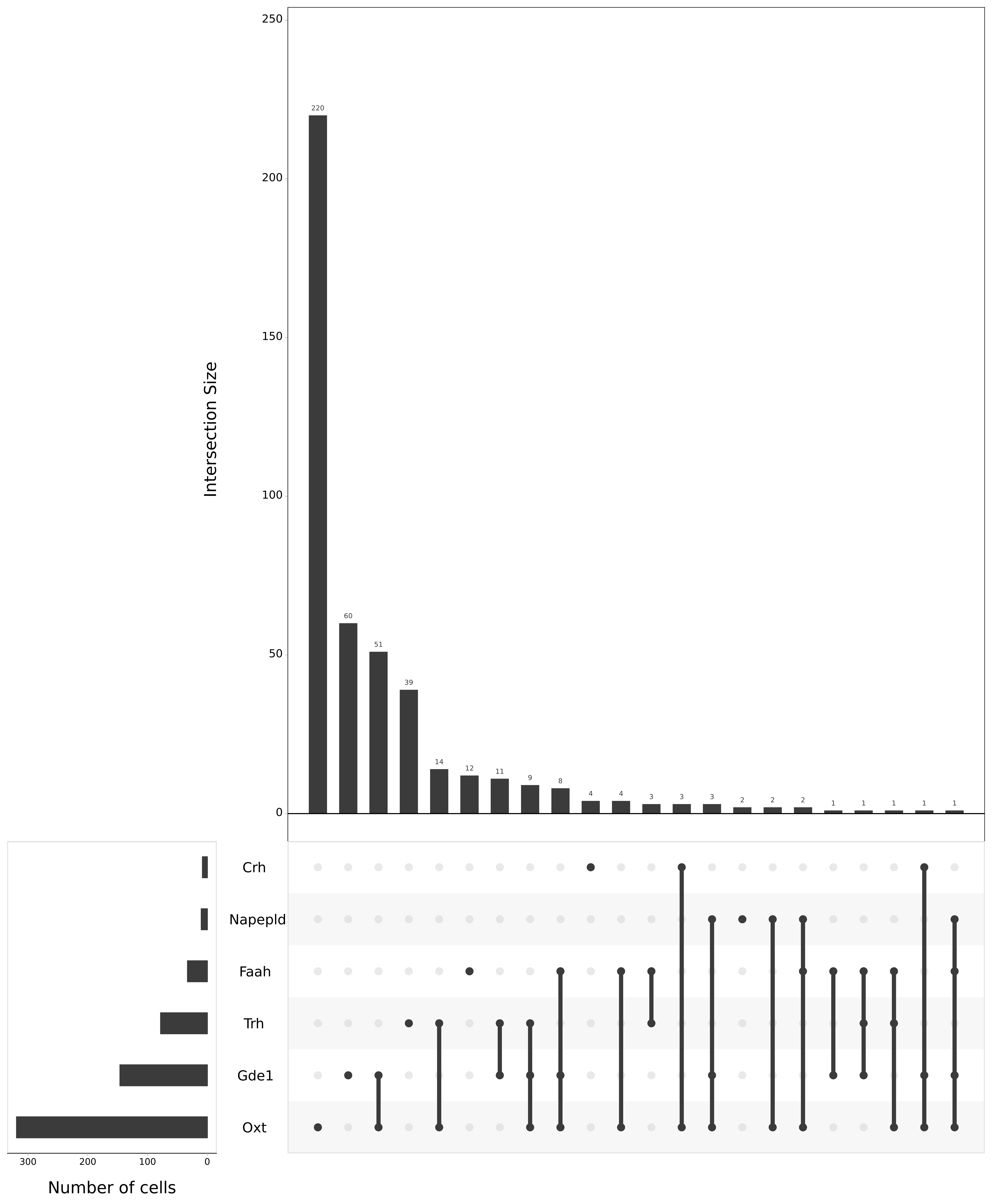
Rows: 51,199
Columns: 38
$ Slc17a6 <dbl> 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,…
$ Slc17a8 <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,…
$ Slc1a1 <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,…
$ Slc1a2 <dbl> 0, 1, 0, 1, 0, 1, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0,…
$ Slc1a6 <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,…
$ Gad1 <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0,…
$ Slc32a1 <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,…
$ Slc6a1 <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0,…
$ Cnr1 <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,…
$ Gpr55 <dbl> 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,…
$ Dagla <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,…
$ Daglb <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,…
$ Mgll <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,…
$ Faah <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,…
$ Napepld <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,…
$ Gde1 <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0,…
$ Pparg <dbl> 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,…
$ nGene <int> 1652, 782, 447, 1706, 1106, 894, 727, 734, 669, 617, …
$ nUMI <dbl> 2787, 1090, 544, 2709, 1817, 1220, 995, 1036, 920, 86…
$ orig.ident <fct> Hypothalamus, Hypothalamus, Hypothalamus, Hypothalamu…
$ res.0.2 <chr> "23", "23", "23", "23", "23", "23", "23", "23", "23",…
$ res.0.4 <chr> "34", "34", "34", "34", "34", "34", "34", "34", "34",…
$ res.0.8 <chr> "42", "42", "42", "42", "42", "42", "42", "42", "42",…
$ res.1.2 <chr> "47", "47", "47", "47", "47", "47", "47", "47", "47",…
$ res.2 <chr> "54", "54", "54", "54", "54", "54", "54", "54", "54",…
$ tree.ident <int> 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1,…
$ pro_Inter <chr> "41", "41", "41", "41", "41", "41", "41", "41", "41",…
$ pro_Enter <chr> "41", "41", "41", "41", "41", "41", "41", "41", "41",…
$ tree_final <fct> 19, 19, 19, 19, 19, 19, 19, 19, 19, 19, 19, 19, 19, 1…
$ subtree <fct> 41, 41, 41, 41, 41, 41, 41, 41, 41, 41, 41, 41, 41, 4…
$ prim_walktrap <fct> 38, 38, 38, 38, 38, 38, 38, 38, 38, 38, 38, 38, 38, 3…
$ umi_per_gene <dbl> 1.687046, 1.393862, 1.217002, 1.587925, 1.642857, 1.3…
$ log_umi_per_gene <dbl> 0.22712693, 0.14421974, 0.08529138, 0.20082998, 0.215…
$ nCount_RNA <dbl> 2787, 1090, 544, 2709, 1817, 1220, 995, 1036, 920, 86…
$ nFeature_RNA <int> 1652, 782, 447, 1706, 1106, 894, 727, 734, 669, 617, …
$ wtree <fct> 38, 38, 38, 38, 38, 38, 38, 38, 38, 38, 38, 38, 38, 3…
$ age <chr> "P23", "P02", "P02", "P02", "P02", "P02", "P02", "P02…
$ stage <ord> Adult, Neonatal, Neonatal, Neonatal, Neonatal, Neonat…