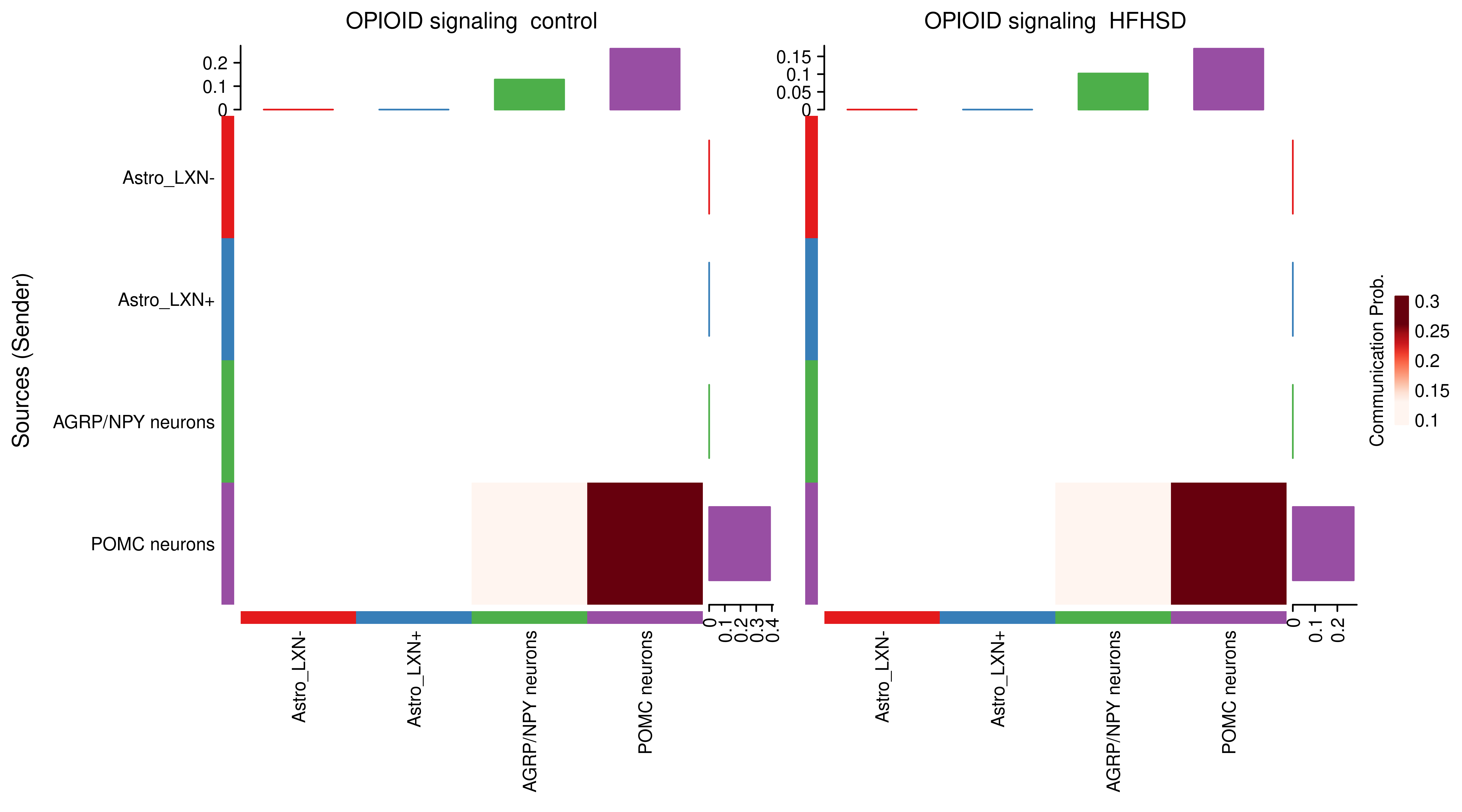
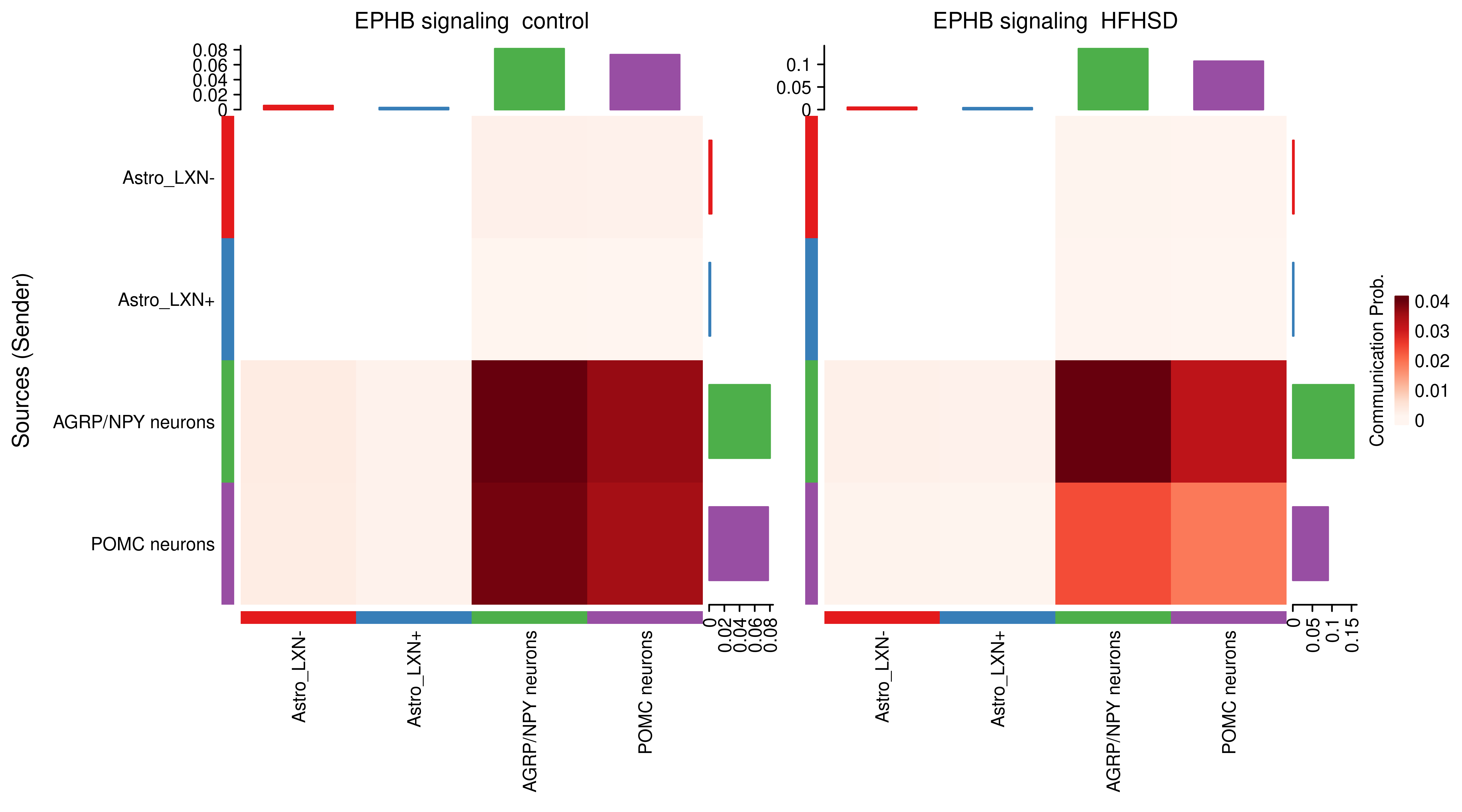
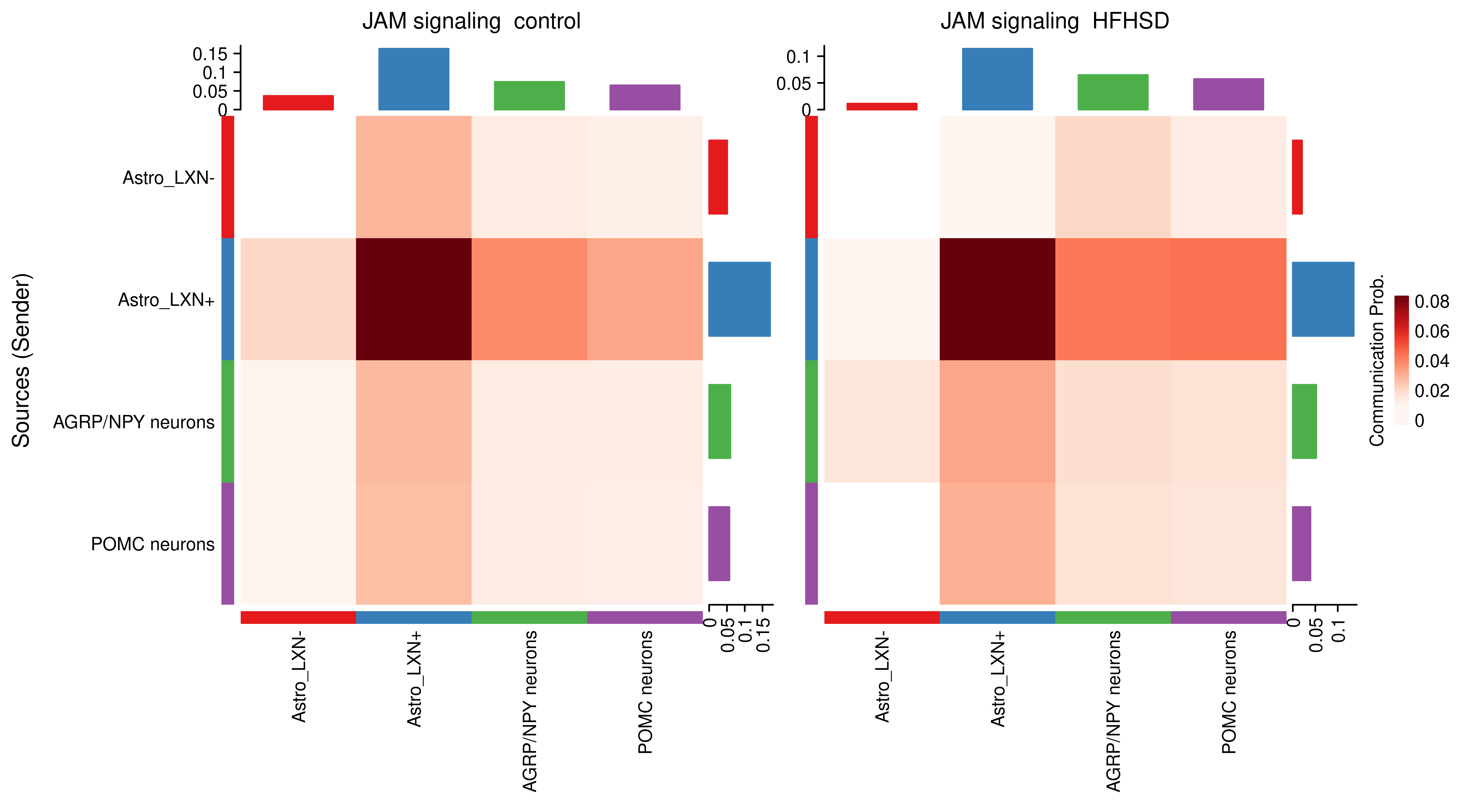
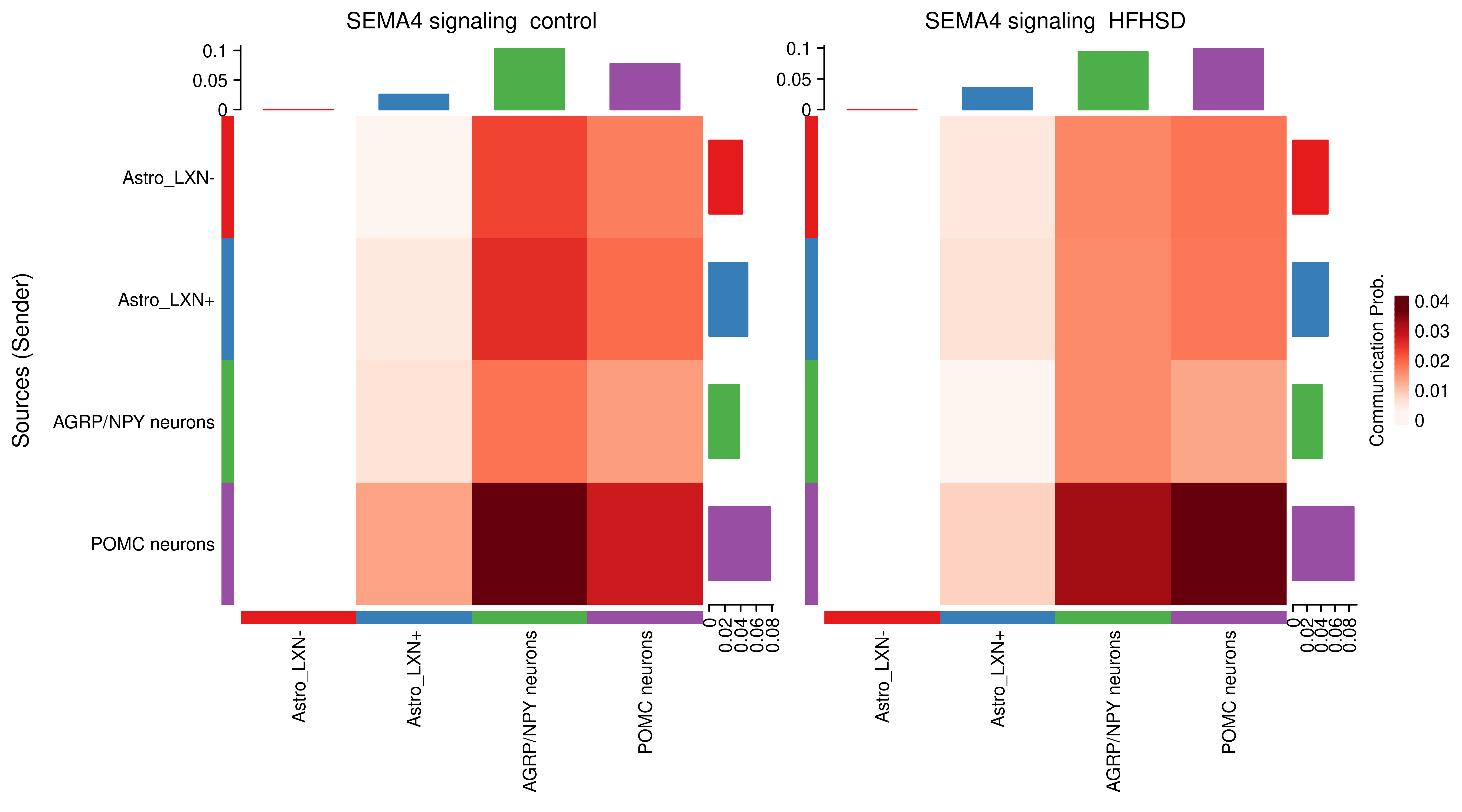
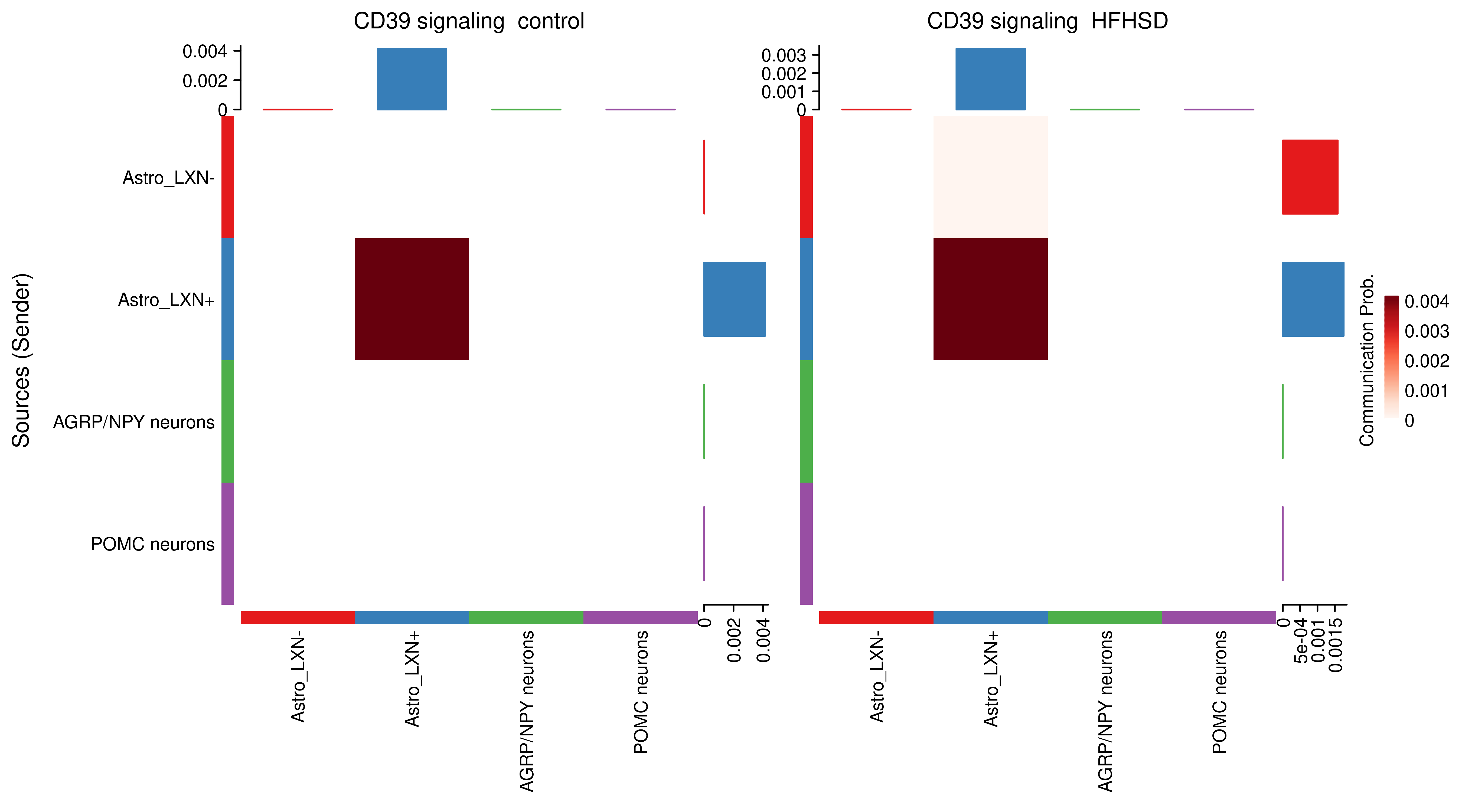
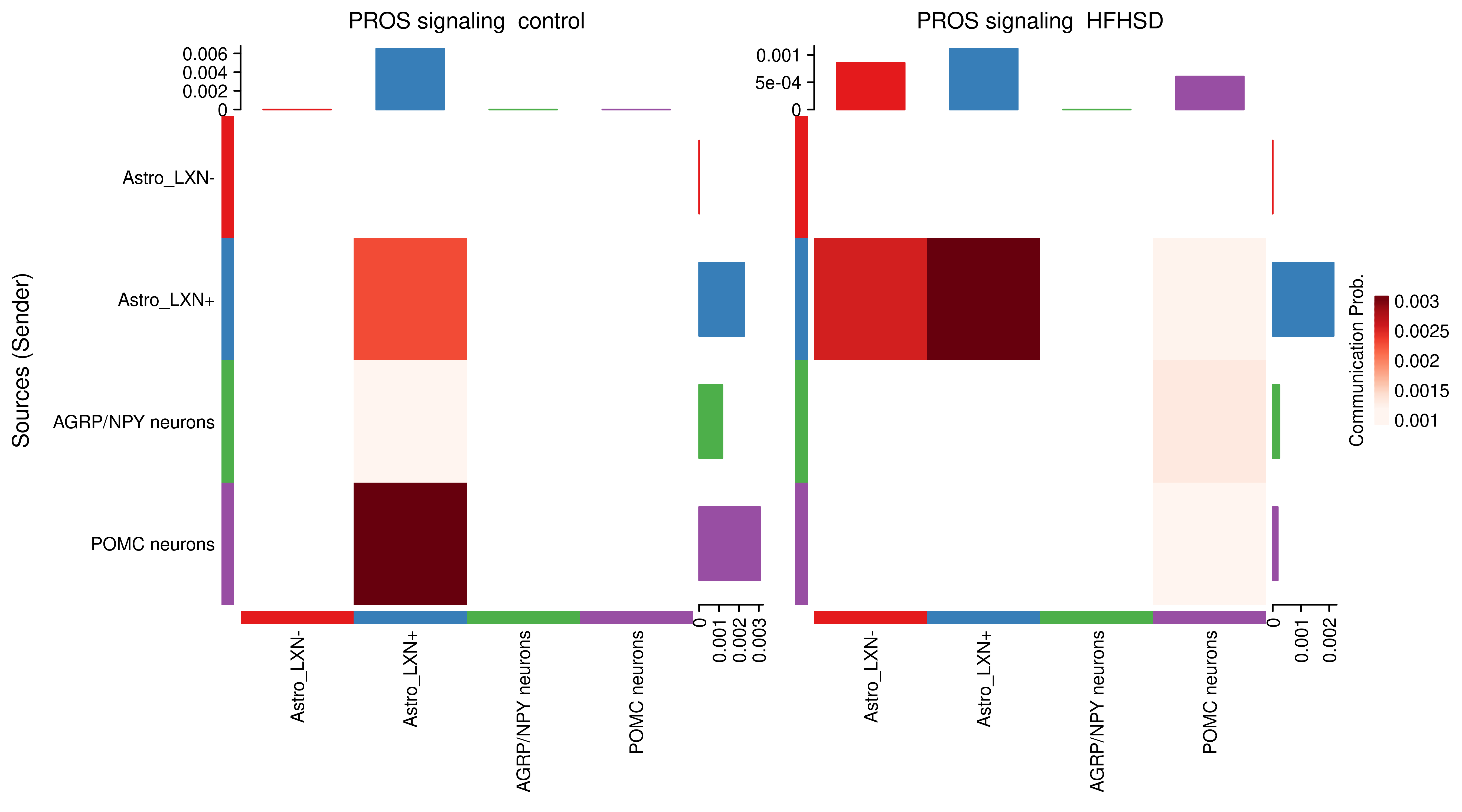
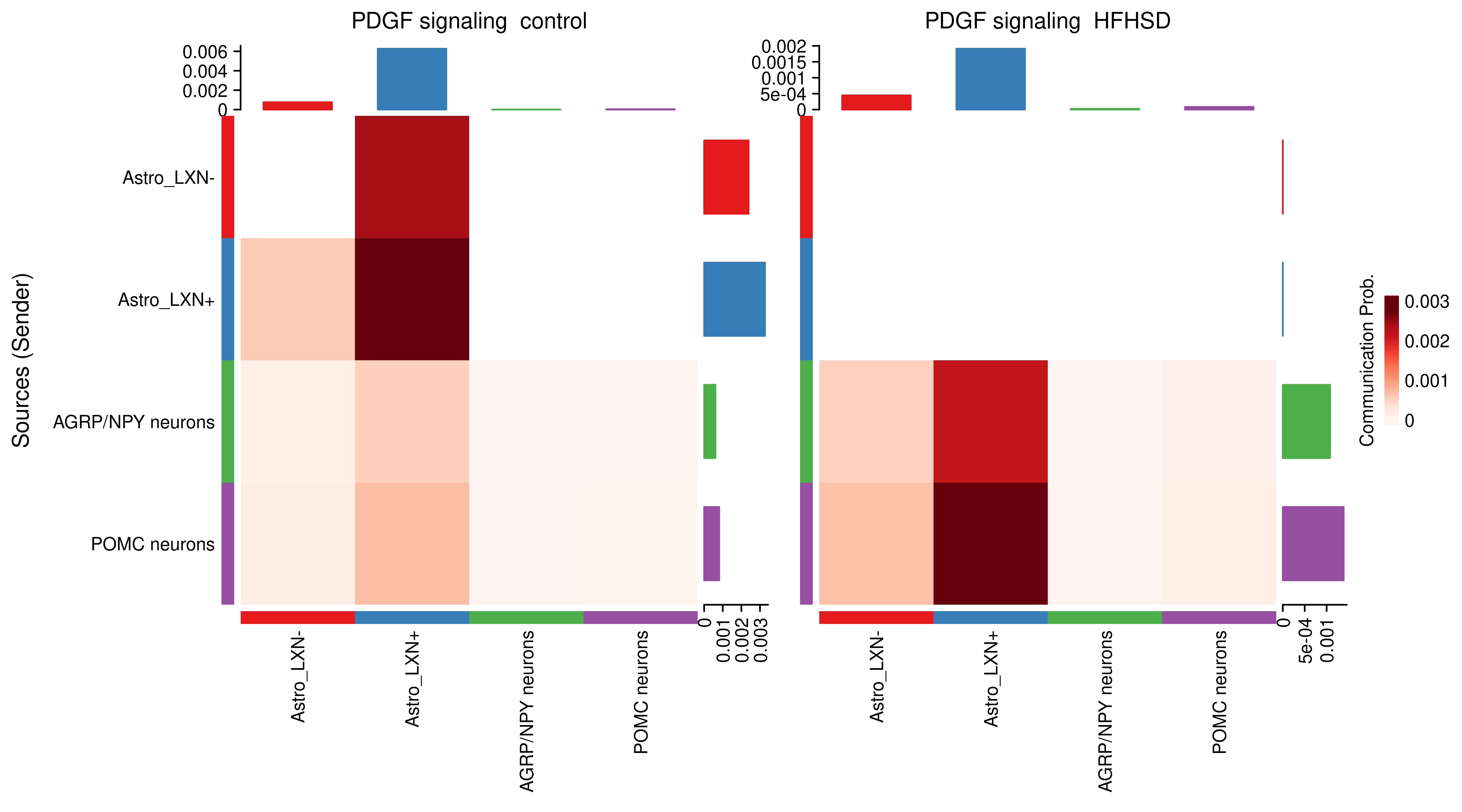
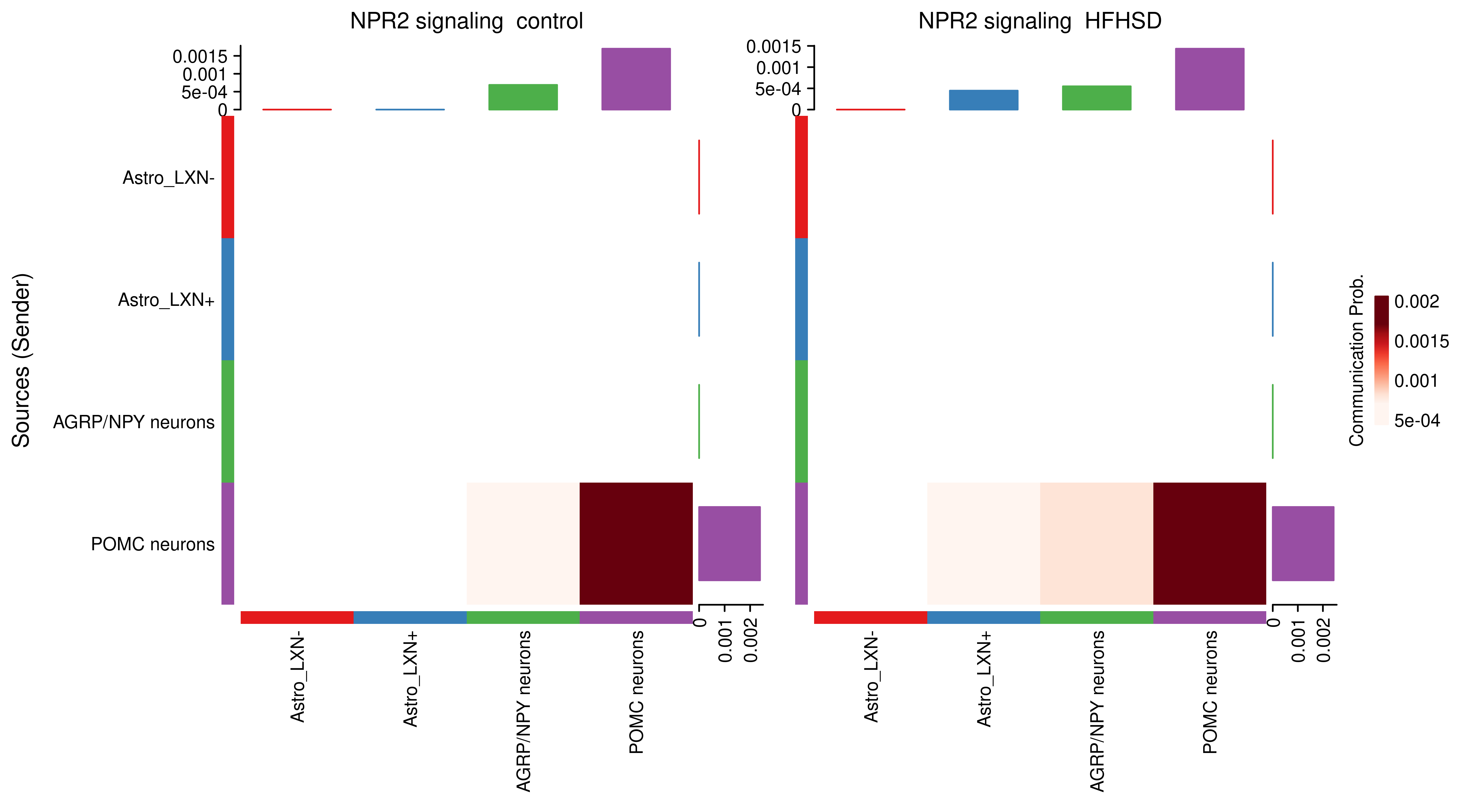
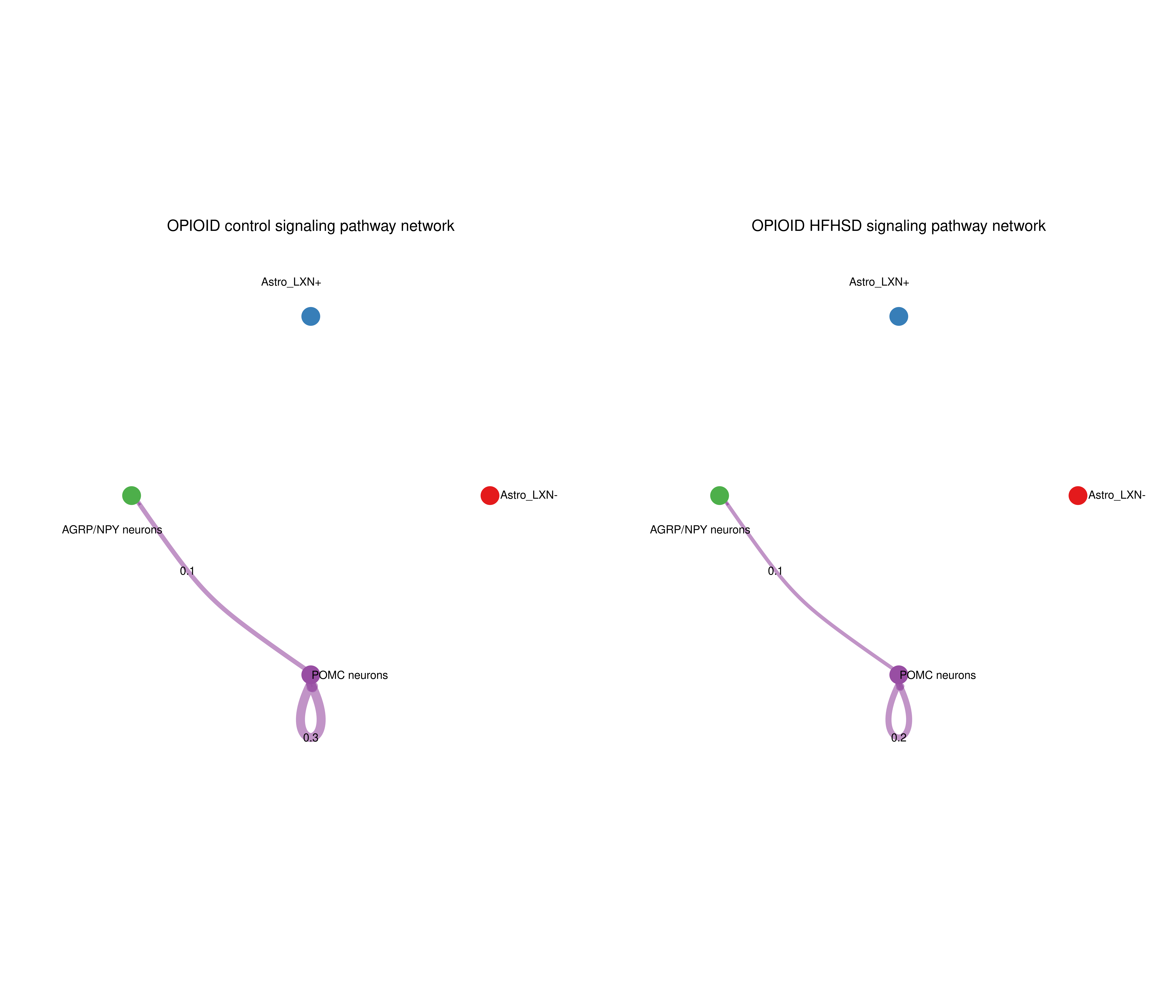
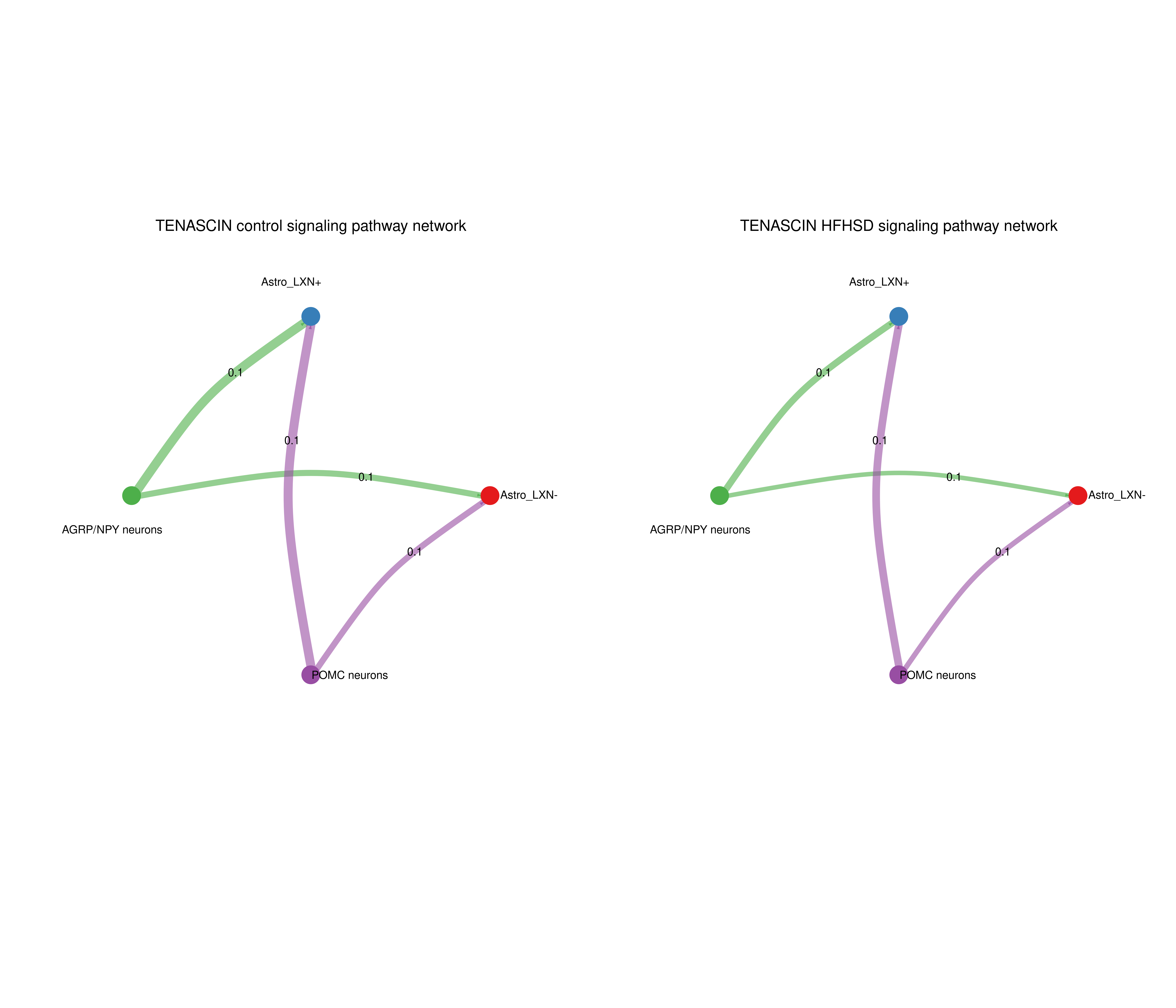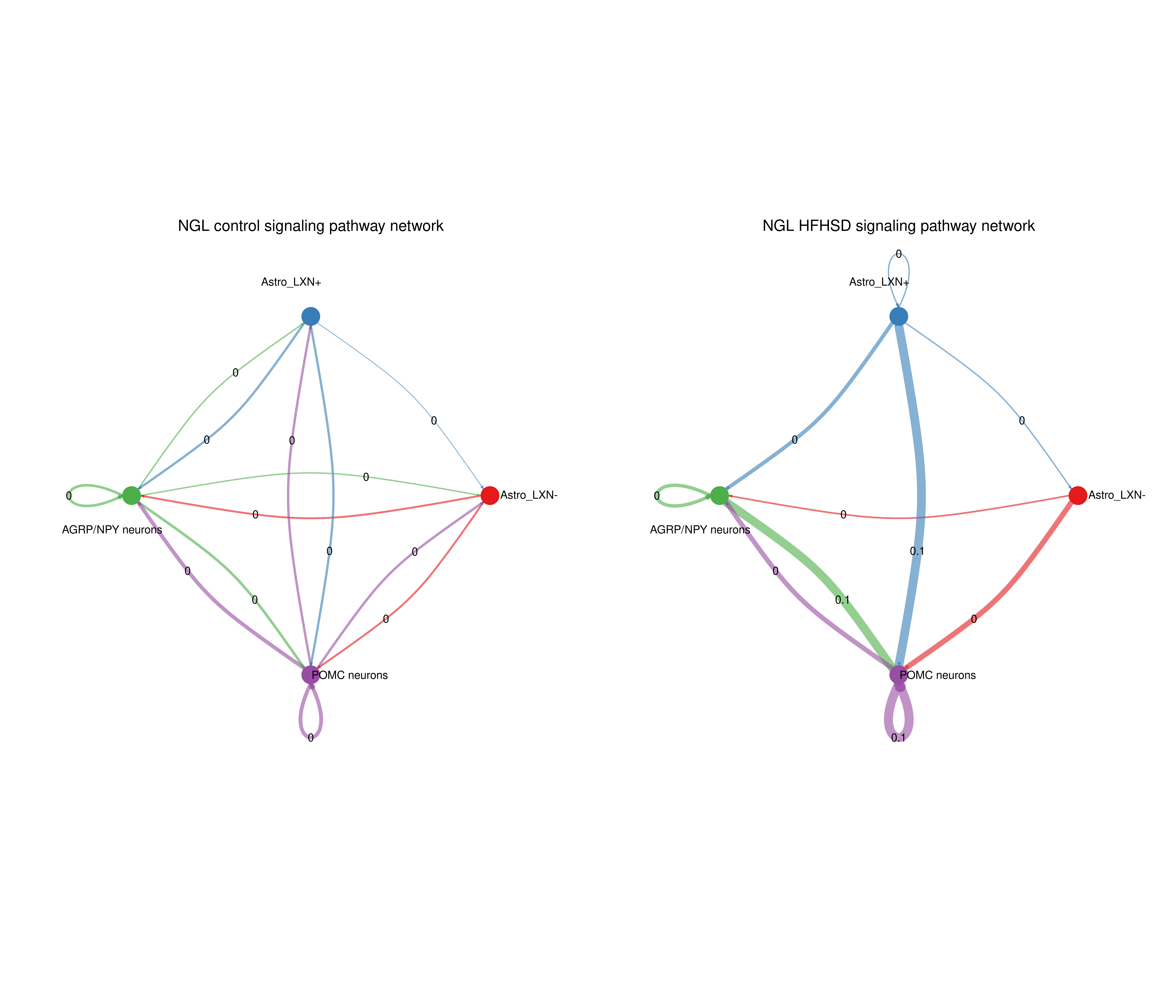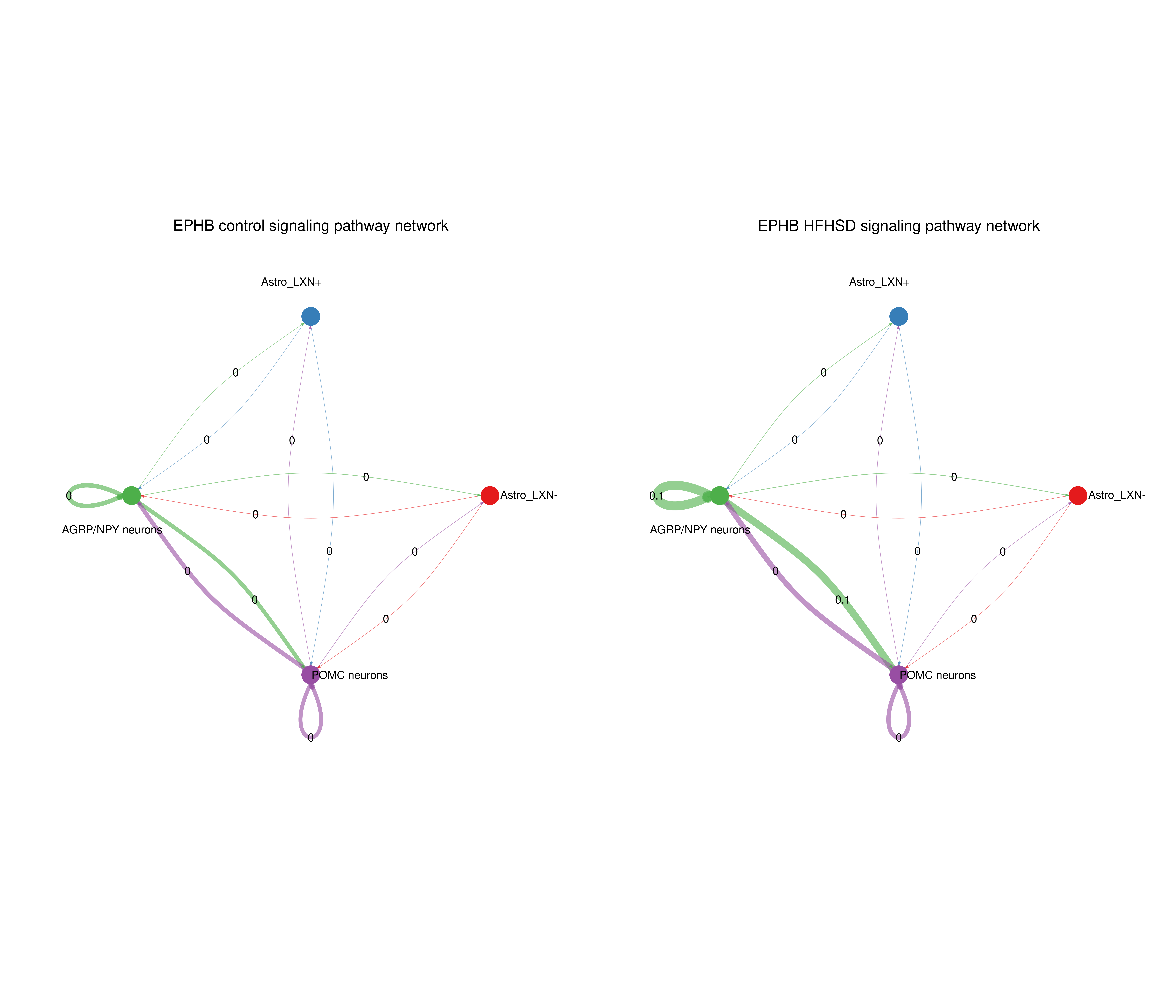AnnData object with n_obs × n_vars = 14594 × 24746
obs: 'nCount_RAW', 'nFeature_RAW', 'nCount_RNA', 'nFeature_RNA', 'orig.ident', 'nFeature_Diff', 'nCount_Diff', 'percent_mito', 'percent_ribo', 'percent_mito_ribo', 'percent_hb', 'log10GenesPerUMI', 'cell_name', 'barcode', 'latent_RT_efficiency', 'latent_cell_probability', 'latent_scale', 'doublet_score', 'predicted_doublets', 'QC', 'var_regex', 'RNA_snn_res.0.5', 'RNA_snn_res.0.71728703859461', 'RNA_snn_res.1.18939286532015', 'RNA_snn_res.3.00000100000001', 'seurat_clusters', 'k_tree', 'comb_clstr1', 'S.Score', 'G2M.Score', 'Phase', 'nCount_SCT', 'nFeature_SCT', 'SCT_snn_res.1', 'SCT_snn_res.1.10031502002514', 'SCT_snn_res.1.2198400795032', 'SCT_snn_res.1.36467746508232', 'SCT_snn_res.1.54382016547197', 'SCT_snn_res.1.77109349840298', 'SCT_snn_res.2.0689045782524', 'SCT_snn_res.2.47612417203789', 'SCT_snn_res.3.06661007679595', 'SCT_snn_res.4.00000100000002', 'bioproject', 'project', 'model', 'tech', 'region', 'sex', 'stage', 'libname', 'expbtch', 'condit', 'ora_celltype'
var: 'vst.mean', 'vst.variance', 'vst.variance.expected', 'vst.variance.standardized', 'vst.variable'
uns: 'k_tree_colors', 'name', 'ora_celltype_colors'
obsm: 'X_pacmap', 'X_pca', 'X_umap', 'ora_estimate', 'ora_pvals'